Download presentation
Presentation is loading. Please wait.

1
Lecture 1 Introduction to recombinant DNA Technology Dr Muhammad Imran

2
What is a gene? Gene is a piece of DNA which encode an RNA molecule which may encode a protein What is a genetic engineering? Set of techniques by which one can deliberately insert new piece/s of DNA into the existing DNA piece to modify the characters of an organism. Gene Cloning Set of experiments carried out to create a recombinant molecule and its propagation in an organism/host organism multiplication.

3
PCR: Polymerase Chain Reaction A reaction in which we use DNA polymerase to make the copies of fragment of DNA selectively amplified with the help of primers

4
History of rDNA Technology Gregor Mendel1850s and 1860s the birth of genetics

5
What genes are and how they work W. Sutton…the factors (genes) reside on Chromosomes 1903. TH Morgan ……. Endorsed Sutton…..and gene mapping started in 1910 and by 1922 nearly 2000 genes were mapped. Set of experiments by Avery, MacLeod, and McCarty in 1944, and of Hershey and Chase in 1952 proved that DNA is hereditary material and not the proteins

6
1952-1966 well-done Watson and Crick Structure of DNA was elucidated, genetic code cracked, and the processes of transcription and translation described Anticlimax era and frustration in late 1960 1971–1973 recombinant DNA technology or genetic engineering Gene cloning Kary Mullis discovered a revolutionary technique now called PCR

7
Gene Cloning T.A Brown 6 th Edition

8
Properties to DNA and its replication DNA is double helix Double helix is anti-parallel Replication only takes place from 5-3 Replication is semi conservative Replication is bidirectional

9
PCR: Polymerase Chain Reaction Quite different from gene cloning Very simple Easy to do less time consuming Economical Wide application

10
PCR temperature profile

11
Critical temperature

12
Melting temperature or Tm of Primers Melting Temperature or Tm. The Tm is the temperature at which the correctly base-paired hybrid dissociates (“melts”). Tm = (4 × [G + C]) + (2 × [A + T])°C TAB
. Tm = (4 × [G + C]) + (2 × [A + T])°C TAB.")
13
Contents of the reaction dNTPs Tag (enzyme) Buffer Primers F and R Template MgCl 2 (NH 4 ) 2 SO 4 or not KCl
 Buffer Primers F and R Template MgCl 2 (NH 4 ) 2 SO 4 or not KCl")
14
PCR reaction contents https://www.neb.com/protocols/1/01/01/protocol-for-a-routine-taq-pcr-reaction

15
Principle of Primer designing Few things to be considered while designing the primers ParametersOptimumComments Primer Length18-22 Primer Melting Temperature 52-58 o C Primer Annealing Temperature T a = 0.3 x T m (primer) + 0.7 T m (product) – 14.9 GC Content40-60% GC Clamp Primer Secondary Structures Repeats4 dinucleotide repears allowed eg ATATATAT RunsConsecutive single nucleotide repeat of 4 max allowed (otherwise mispriming)
 T m (product) – 14.9 GC Content40-60% GC Clamp Primer Secondary Structures Repeats4 dinucleotide repears allowed eg ATATATAT RunsConsecutive single nucleotide repeat of 4 max allowed (otherwise mispriming)")
16
Continued……………….. ParametersOptimumComments 3' End Stability Avoid Template Secondary Structure Avoid Cross Homology

17
Primer for different purposes Simple primer (Universal) Degenerate primers ARMS PCR Primer Multiplex PCR primers Primers for protein expression Primers for site directed mutagenesis When we need them?
 Degenerate primers ARMS PCR Primer Multiplex PCR primers Primers for protein expression Primers for site directed mutagenesis When we need them")
18
Simple Primer (universal primers) When we want to amplify a region for sequencing, homologous sequences are available to design primers in large number. 16S rDNA primers (Universal primer) When large data of identical sequences is known ClCuD universal primers Universal primers for sequencing clones in expression vectors or TA cloning vectors etc. T7 promoter forward: TAATACGACTCACTATAGGG T7 terminator reverse: GCTAGTTATTGCTCAGCGG http://www.addgene.org/mol_bio_reference/sequencing_primers/
 When large data of identical sequences is known ClCuD universal primers Universal primers for sequencing clones in expression vectors or TA cloning vectors etc. T7 promoter forward: TAATACGACTCACTATAGGG T7 terminator reverse: GCTAGTTATTGCTCAGCGG")
19
Degenerate Primers When the polymorphism in region to be amplified exist. When primers have to be designed from protein sequence or conserved protein domain

20
ACGTA/CA/gA/TC/gC/Tg/TA/C/gA/C/TA/g/TC/g/TA/C/g/T ACGTMRWSYKVHDBN

21
Degenerate primers cont……141 ACGTA/CA/gA/TC/gC/Tg/TA/C/gA/C/TA/g/TC/g/TA/C/g/T ACGTMRWSYKVHDBN F Primer 5’ACNgARgCNCARTAYgAR ATg3’

22
Reverse degenerate primer 233 ACGTA/CA/gA/TC/gC/Tg/TA/C/gA/C/TA/g/TC/g/TA/C/g/T ACGTMRWSYKVHDBN

23
ARMS (Amplification refractory mutation system) Watch out, outer primer colors are bright and light you may have difficulty in seeing them
 Watch out, outer primer colors are bright and light you may have difficulty in seeing them")
24
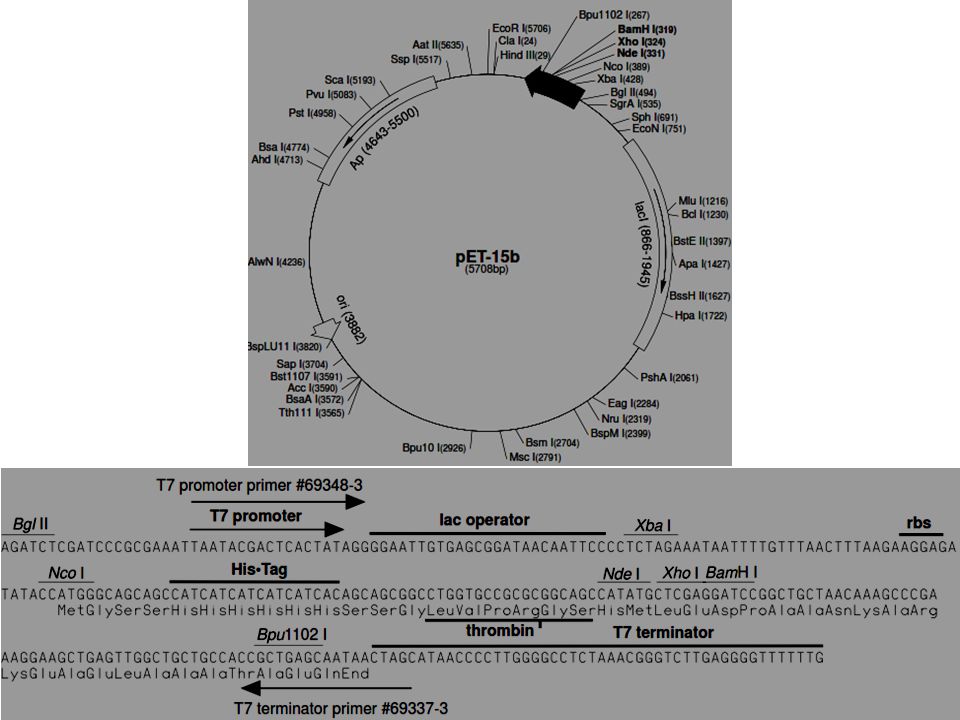
Primers for protein expression

25
Restriction site. Should be same as for vector or produce same sticky ends as of 1 st site in MCS Blunt end cutter Nco I CCATGG) or Nde I (CATATG) are mostly chosen as the ATG within these sites can be used directly to create the ATG start codon and/or the ATG codon for the N-terminal methionine residue. Design of the 5'-end primer Following modification can be made to the 5 prime end of a primer
 or Nde I (CATATG) are mostly chosen as the ATG within these sites can be used directly to create the ATG start codon and/or the ATG codon for the N-terminal methionine residue. Design of the 5 -end primer Following modification can be made to the 5 prime end of a primer.")
26
5'-extension to the restriction site http://www.embl.de/pepcore/pepcore_services/cloning/pcr_strategy/primer_design/extensions.pdf

27
Design of the 5'-end primer Start codon. 1) If no N terminal tag/fusion partner….include a start codon (usually ATG). 2)- or if an N-terminal methionine residue is present. 3) Make sure that N terminal tag/fusion partner is in frame with gene of interest Overlap with the gene of interest. The overlap between the primer and the gene of interest should be long enough to give a T m of 60°C or more
 If no N terminal tag/fusion partner….include a start codon (usually ATG). 2)- or if an N-terminal methionine residue is present. 3) Make sure that N terminal tag/fusion partner is in frame with gene of interest Overlap with the gene of interest. The overlap between the primer and the gene of interest should be long enough to give a T m of 60°C or more.")
29
Design of the 3'-end primer Restriction site. Should be same as for vector or produce same sticky ends as of 2 nd or 3 rd site in MCS or use blunt end cutter Stop codon(s). If no C-terminal tag introduce a stop codon TAA is preferred over TAG/TGA Two or three stop codon sequences can be incorporated for efficient termination Overlap with the gene of interest. The overlap between the primer and the gene of interest should be long enough to give a T m of 60°C or more
. If no C-terminal tag introduce a stop codon TAA is preferred over TAG/TGA Two or three stop codon sequences can be incorporated for efficient termination Overlap with the gene of interest. The overlap between the primer and the gene of interest should be long enough to give a T m of 60°C or more.")
30
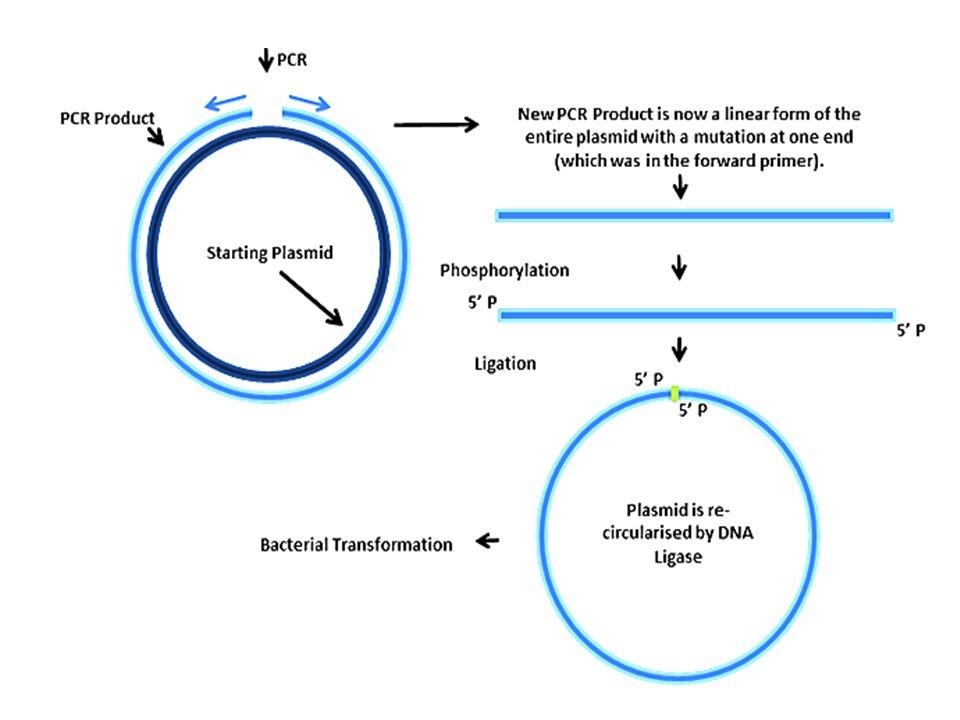
Primers for site directed mutagenesis

31
Back to back primer design (principle) Incorporate the desired mutation at the 5 end of a primer or both primers We usually insert the mutation in the middle of the primer (better result, draw on board) Calculate the Tm considering mismatches http://depts.washington.edu/bakerpg/primertemp/primertemp.html
 Incorporate the desired mutation at the 5 end of a primer or both primers We usually insert the mutation in the middle of the primer (better result, draw on board) Calculate the Tm considering mismatches")
32
http://oxfordgenetics.com/cloning-resources/cloning-guides/site-directed-mutagenesis

Similar presentations







 Analysis of DNA (Sequencing) Chemical Synthesis.>")


 Analysis of DNA (Sequencing) Chemical Synthesis of DNA.>")








