Download presentation
Presentation is loading. Please wait.

1
Terapia chirurgica della cardiopatia ischemica
Anatomia e fisiopatologia del circolo coronarico

2
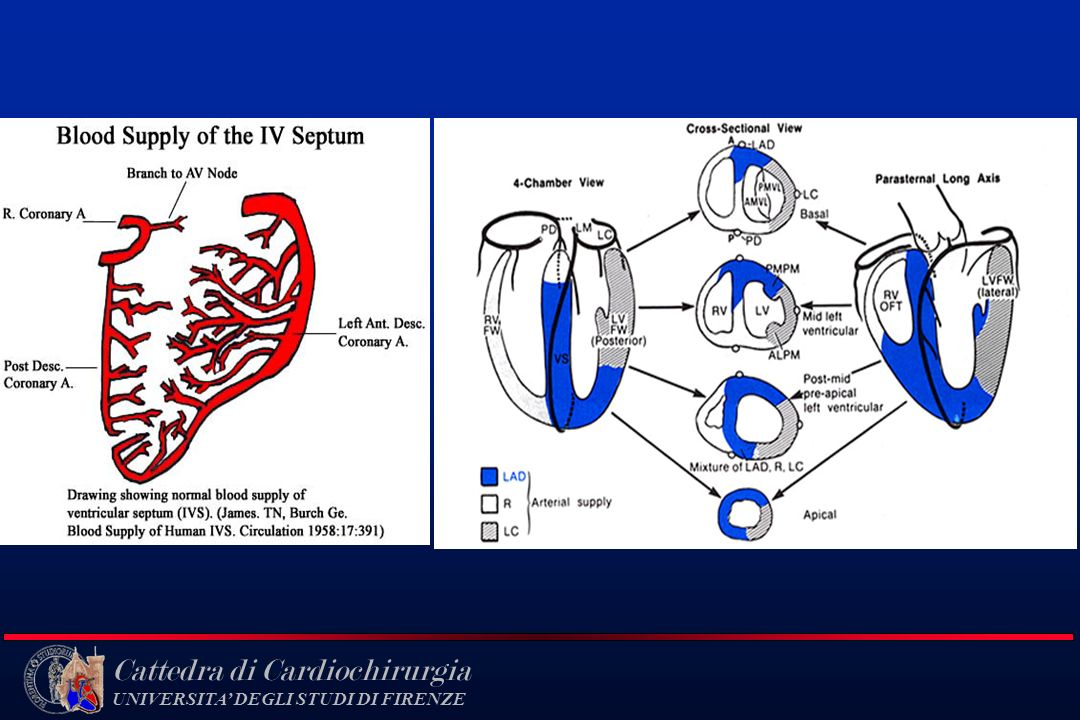
Anatomia Coronarica

7
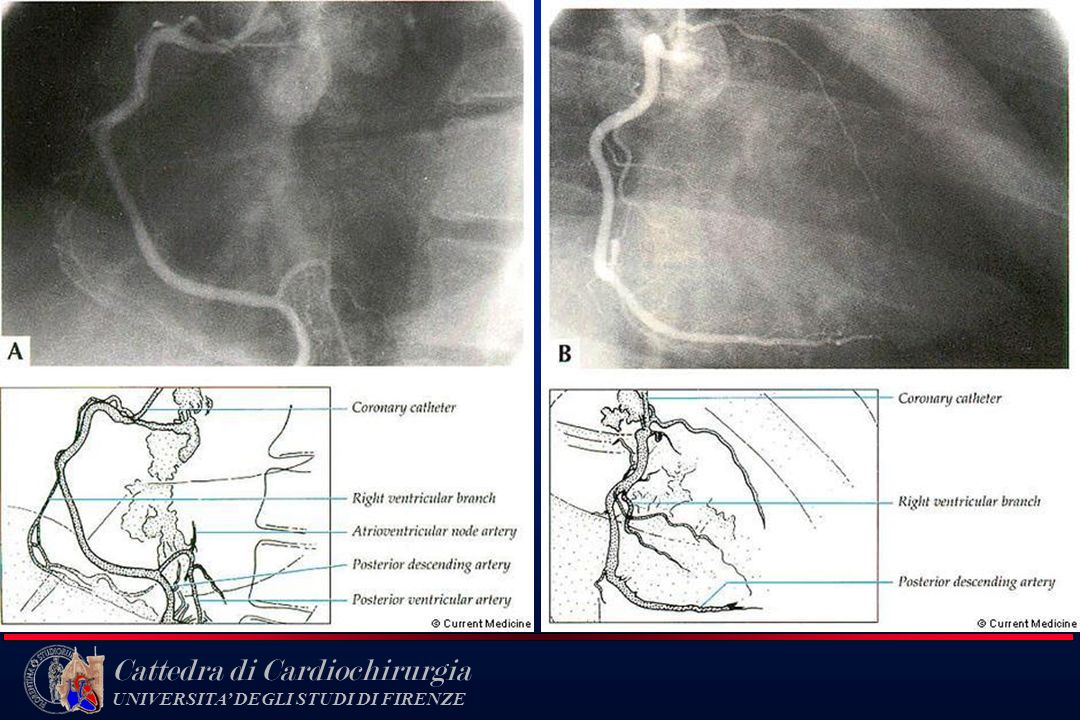
Coronarografia

10
Electron-Beam CT Images of the Heart

11
CORONARY CIRCULATION Blood flow
left ventricle = 80/ml/min/100g right ventricle = 40 ml/min/100g atria = 20 ml/min/100g *Flow can increase 4-fold Capillary density - all capillaries open Very high O2 extraction: (A-V)02 = 14 ml 02/dl VO2 = 12 ml/min/100g ----> very high
02 = 14 ml 02/dl. VO2 = 12 ml/min/100g ----> very high.")
12
MYOCARDIAL OXYGEN CONSUMPTION
a to cardiac work influenced by a) contractility b) heart rate c) after-load increases achieved primarily by hyperemia 40% due to oxidation of carbohydrates, 60% fatty acids
 contractility. b) heart rate. c) after-load. increases achieved primarily by hyperemia. 40% due to oxidation of carbohydrates, 60% fatty acids.")
13
TRANSMURAL DISTRIBUTION OF BLOOD FLOW
contraction (systole) leads to compression of intramural vessels and reduction in flow pressure inside left ventricle can exceed aortic pressure during systole vessel compression greatest in endocardium, decreases toward epicardium O2 demand and flow/g is greatest in endocardium LV coronary flow decreases as HR increases since diastole shorter
 leads to compression of intramural vessels and reduction in flow. pressure inside left ventricle can exceed aortic pressure during systole. vessel compression greatest in endocardium, decreases toward epicardium. O2 demand and flow/g is greatest in endocardium. LV coronary flow decreases as HR increases since diastole shorter.")
14
Perfusione coronarica “fasica”
150 Systole Diastole Intramyocardial Pressure (mmHg) 100 Arterial Blood Pressure 120 100 50 80 Left Coronary Blood Flow 150 Zone flow 100 Left Ventricular Pressure (mmHg) Right Coronary Blood Flow 50 Zero Flow Perfusione coronarica “fasica”
 100. Arterial Blood. Pressure Left Coronary. Blood Flow Zone flow Left Ventricular Pressure (mmHg) Right Coronary. Blood Flow. 50. Zero Flow. Perfusione coronarica fasica")
15
HEART RATE AND CORONARY BLOOD FLOW
time in systole vessel compression Tachycardia: HR metabolic activity vasodilation time in systole vessel compression Bradycardia: HR metabolic activity vasoconstriction

16
Oxygen Consumption (ml / 100 gm / min)
. 18 16 14 12 Oxygen Consumption (ml / 100 gm / min) 10 8 6 4 2 20 30 40 50 60 70 80 90 100 110 120 Coronary Flow (ml / 100 gm / min)
 Coronary Flow (ml / 100 gm / min)")
17
LOCAL CONTROL OF CORONARY BLOOD FLOW
Tissue oxygenation is major regulator of vascular tone (adenosine, tiss pO2) Essentially all capillaries are open to flow (O2 diffusion distance) Flow regulation occurs at arterioles VO2 limited by blood flow (max O2 extraction)
 Essentially all capillaries are open to flow (O2 diffusion distance) Flow regulation occurs at arterioles. VO2 limited by blood flow (max O2 extraction)")
19
Endothelial Dysfunction in Atherosclerosis
The earliest changes that precede the formation of lesions of atherosclerosis take place in the endothelium. These changes include increased endothelial permeability to lipoproteins and other plasma constituents, which is mediated by nitric oxide, prostacyclin, platelet-derived growth factor, angiotensin II, and endothelin; up- regulation of leukocyte adhesion molecules, including L-selectin, integrins, and platelet–endothelial-cell adhesion molecule 1, and the up-regulation of endothelial adhesion molecules, which include E-selectin, P-selectin, intercellular adhesion molecule 1, and vascular-cell adhesion molecule 1; and migration of leukocytes into the artery wall, which is mediated by oxidized low-density lipoprotein, monocyte chemotactic protein 1, interleukin-8, platelet-derived growth factor, macrophage colony-stimulating factor, and osteopontin.

20
Fatty-Streak Formation in Atherosclerosis
Fatty streaks initially consist of lipid-laden monocytes and macrophages (foam cells) together with T lymphocytes. Later they are joined by various numbers of smooth-muscle cells. The steps involved in this process include smooth-muscle migration, which is stimulated by platelet-derived growth factor, fibroblast growth factor 2, and transforming growth factor b; T-cell activation, which is mediated by tumor necrosis factor a, interleukin-2, and granulocyte–macrophage colony-stimulating factor; foamcell formation, which is mediated by oxidized low-density lipoprotein, macrophage colony-stimulating factor, tumor necrosis factor a, and interleukin-1; and platelet adherence and aggregation, which are stimulated by integrins, P-selectin, fibrin, thromboxane A2, tissue factor, and the factors described as responsible for the adherence and migration of leukocytes.
 together with T lymphocytes. Later they are joined by various numbers of smooth-muscle cells. The steps involved in this process include smooth-muscle migration, which is stimulated by platelet-derived growth factor, fibroblast growth factor 2, and transforming growth factor b; T-cell activation, which is mediated by tumor necrosis factor a, interleukin-2, and granulocyte–macrophage colony-stimulating factor; foamcell formation, which is mediated by oxidized low-density lipoprotein, macrophage colony-stimulating factor, tumor necrosis factor a, and interleukin-1; and platelet adherence and aggregation, which are stimulated by integrins, P-selectin, fibrin, thromboxane A2, tissue factor, and the factors described as responsible for the adherence and migration of leukocytes.")
21
Formation of an Advanced, Complicated Lesion of Atherosclerosis
As fatty streaks progress to intermediate and advanced lesions, they tend to form a fibrous cap that walls off the lesion from the lumen. This represents a type of healing or fibrous response to the injury. The fibrous cap covers a mixture of leukocytes, lipid, and debris, which may form a necrotic core. These lesions expand at their shoulders by means of continued leukocyte adhesion and entry The principal factors associated with macrophage accumulation include macrophage colony-stimulating factor, monocyte chemotactic protein 1, and oxidized low-density lipoprotein. The necrotic core represents the results of apoptosis and necrosis, increased proteolytic activity, and lipid accumulation. The fibrous cap forms as a result of increased activity of platelet-derived growth factor, transforming growth factor b, interleukin-1, tumor necrosis factor a , and osteopontin and of decreased connective-tissue degradation.

22
Unstable Fibrous Plaques in Atherosclerosis
Rupture of the fibrous cap or ulceration of the fibrous plaque can rapidly lead to thrombosis and usually occurs at sites of thinning of the fibrous cap that covers the advanced lesion. Thinning of the fibrous cap is apparently due to the continuing influx and activation of macrophages, which release metalloproteinases and other proteolytic enzymes at these sites. These enzymes cause degradation of the matrix, which can lead to hemorrhage from the vasa vasorum or from the lumen of the artery and can result in thrombus formation and occlusion of the artery.

23
Pathophysiologic Events Culminating in the Clinical Syndrome of Unstable Angina
Numerous physiologic triggers probably initiate the rupture of a vulnerable plaque. Rupture leads to the activation, adhesion, and aggregation of platelets and the activation of the clotting cascade, resulting in the formation of an occlusive thrombus. If this process leads to complete occlusion of the artery, then acute myocardial infarction with ST-segment elevation occurs. Alternatively, if the process leads to severe stenosis but the artery nonetheless remains patent, then unstable angina occurs.

24
Atheroma morphology by intravascular ultrasound (IVUS)
")
27
Atherosclerosis Timeline
Foam Cells Fatty Streak Intermediate Lesion Atheroma Fibrous Plaque Complicated Lesion / Rupture Atherosclerosis is a progressive disease involving the development of arterial wall lesions. As they grow, these lesions may narrow or occlude the arterial lumen. Complex lesions may also become unstable and rupture, leading to acute coronary events, such as unstable angina, myocardial infarction, and stroke. Pepine CJ. The effects of angiotensin-converting enzyme inhibition on endothelial dysfunction: potential role in myocardial ischemia. Am J Cardiol. 1998; 82(suppl 10A): Endothelial Dysfunction From First Decade From Third Decade From Fourth Decade Adapted from Pepine CJ. Am J Cardiol. 1998;82(suppl 104). 9
: Endothelial Dysfunction. From First. Decade. From Third. Decade. From Fourth. Decade. Adapted from Pepine CJ. Am J Cardiol. 1998;82(suppl 104). 9.")
30
CHD Narrowing of Coronary artery limits blood supply to heart muscle
If demand for blood supply cannot be met, muscle becomes ischaemic Narrowing of Coronary artery limits blood supply to heart muscle

31
The Three Possible Outcomes of Myocardial Ischemia
3) Relief of ischemia 1) Myocardial infarction 2) Chronic Ischemia without infarction Salvage of previously ischemic myocardium Hearts with elements of both hibernating and stunning: Transient postischemic dysfunction: Persistent Ischemia dysfunction: Stunned/ Hibernating myocardium Hibernating myocardium Stunned myocardium ? ? Relief of ischemia No return of contractile function Return of contractile function Adapted from Kloner, R., et al., Myocardial stunning and hibernation: Mechanisms and clinical implications. In Braunwald, E. (ed.): Heart Disease: A textbook of Cardiovascular Medicine, 3rd ed. Philadelphia, W.B., Aaunders Company. Update No. 11, p. 253, 1990.
 Relief of ischemia. 1) Myocardial. infarction. 2) Chronic Ischemia. without infarction. Salvage of previously. ischemic myocardium. Hearts with elements. of both hibernating. and stunning: Transient. postischemic. dysfunction: Persistent Ischemia. dysfunction: Stunned/ Hibernating. myocardium. Hibernating myocardium. Stunned myocardium. Relief of ischemia. No return of. contractile function. Return of. contractile function. Adapted from Kloner, R., et al., Myocardial stunning and hibernation: Mechanisms and clinical implications. In Braunwald, E. (ed.): Heart Disease: A textbook of Cardiovascular Medicine, 3rd ed. Philadelphia, W.B., Aaunders Company. Update No. 11, p. 253,")
32
Schematic Diagram of Stunned Myocardium
Clamp Wall motion abnormality Coronary occlusion Wall motion abnormality during occlusion Coronary reperfusion Persistent wall motion abnormality (despite reperfusion and viable myocytes) Return of function Gradual return of function (hours to days) From Kloner, R.A., Am J Med 1986;86:14.
 Return of. function. Gradual return of. function (hours to days) From Kloner, R.A., Am J Med 1986;86:14.")
33
Hibernating Myocardium Atherosclerotic narrowing
Wall motion abnormality Atherosclerotic narrowing Wall motion abnormality due to chronic ischemia without infarction From Kloner, R.A., Am J Med 1986;86:14.

34
Remodeling - Definition
Changes in interstitial, cellular, molecular, and genome expression that results in clinical changes in size, shape, and function of the heart

35
Remodeling Post-IMA Early Remodeling (within 72 hours)
¤ involves expansion of the infarct zone Late Remodeling (beyond 72 hours) ¤ involves the left ventricle globally and is involves the left ventricle globally and is associated with time-dependent dilatation, and the distortion of ventricular shape, and mural hypertrophy. Pfeffer MA et al. Circulation 1990;81: White HD et al. Circulation 1987;76:44-51 Martin G. et al. Circulation 2000;101:
 ¤ involves the left ventricle globally and is involves the left ventricle globally and is associated with time-dependent dilatation, and the distortion of ventricular shape, and mural hypertrophy. Pfeffer MA et al. Circulation 1990;81: White HD et al. Circulation 1987;76: Martin G. et al. Circulation 2000;101:")
36
Post infarction remodeling (PIR)
(PIR) Most studied model of remodeling Begins rapidly within hours of infarction There is variation in post infarction remodeling depending on the time of ischaemia, duration, amount of preconditioning, collaterals, genotype, neuroendocrine status, treatment and response.
 Most studied model of remodeling. Begins rapidly within hours of infarction. There is variation in post infarction remodeling depending on the time of ischaemia, duration, amount of preconditioning, collaterals, genotype, neuroendocrine status, treatment and response.")
37
Left ventricular remodeling after myocardial infarction
During the critical initial hours of MI when acute ischemia progresses to true necrosis, regional systolic dysfunction is already present. However, in this particularly crucial period, measures to restore the balance between O2 demand and delivery can lead to salvage of contractile tissue. Once cell death has occurred, and particularly if there is a transmural infarction involving the ventricular apex, there is a high likelihood that this initially functional distortion of ventricular contour will become structural for infarct expansion. The distorted ventricle undergoes further remodeling as a consequence of heightened wall stress on the remaining viable myocardium, which leads to further cavity enlargement and shape distortion. The latter insidious process is associated with a greater likelihood of cardiovascular morbidity and mortality

38
Processes of PIR Infarct expansion - thinning & regional expansion (in
animals occurs within 1 day) Global function impairment - occurs on day 2 Myocyte lengthening Ventricular wall thinning Inflammation and resorption of necrotic tissue Dilatation and reshaping of LV late expansion Myocyte hypertrophy Late myocyte loss Fibrosis and collagen accumulation in interstitium
 Global function impairment - occurs on day 2. Myocyte lengthening. Ventricular wall thinning. Inflammation and resorption of necrotic tissue. Dilatation and reshaping of LV. late expansion. Myocyte hypertrophy. Late myocyte loss. Fibrosis and collagen accumulation in interstitium.")
39
Haemodynamics Thinning of the infarct area
Compensatory hypertrophy of remaining LV The balance of thinning and hypertrophy determines the wall stress and thus the further dilatation of the heart. Therapy can alter these factors

40
Neurohormonal NA Noradrenaline - initially improves CO
Patients with decreasing levels of NA, ANP post MI had better prognosis.

41
Neurohormonal Angiotensin
AII - increases DNA synthesis in fibroblast and increases cell growth and hypertrophy in response to stretch. Also increase permeability and has cytotoxic effects on myocardium. Aldosterone stimulates fibroblasts in collagen synthesis.

42
Cytokines Interleukins, TNF, endothelins PKC, all mediate the remodeling process Increased levels associated with poorer prognosis Blockage of endothelin in animal models improves remodeling Stimulation of TNF alpha can lead to LV remodeling in animals.

43
Oxidative stress Incomplete understanding of oxidative stress and its role in remodeling beyond that of apoptosis. Seems to alter viability of myocytes in the presence of cytokines.

44
Myocytes Myocytes Decreased numbers - residual myocytes lengthen and hypertrophies. This compensates for the loss of other myocytes. Wall stress leading to cell membrane stretching and local neurohormonal and cytokine environment leads to altered expression of hypertrophy associated genes and increased synthesis of contractile proteins.

Similar presentations
















, fibrous material and.>")





>")





