Download presentation
Presentation is loading. Please wait.

1
BioPerl An Introduction to Perl – by Seung-Yeop Lee XS extension – by Sen Zhang BioPerl Introduction– by Hairong Zhao BioPerl Script Examples – by Tiequan Zhang

2
Part I. An Introduction to Perl by Seung-Yeop Lee

3
What is Perl? Perl is an interpreted programming language that resembles both a real programming language and a shell. A Language for easily manipulating text, files, and processes Provides more concise and readable way to do jobs formerly accomplished using C or shells. Perl stands for Practical Extraction and Report Language. Author: Larry Wall (1986)
.")
4
Why use Perl? Easy to use Basic syntax is C-like Type-”friendly” (no need for explicit casting) Lazy memory management A small amount of code goes a long way Fast Perl has numerous built-in optimization features which makes it run faster than other scripting language. Portability One script version runs everywhere (unmodified).
 Lazy memory management A small amount of code goes a long way Fast Perl has numerous built-in optimization features which makes it run faster than other scripting language. Portability One script version runs everywhere (unmodified)..")
5
Why use Perl? Efficiency For programs that perform the same task (C and Perl), even a skilled C programmer would have to work harder to write code that: Runs as fast as Perl code Is represented by fewer lines of code Correctness Perl fully parses and pre-”compiles” script before execution. Efficiently eliminates the potential for runtime SYNTAX errors. Free to use Comes with source code
, even a skilled C programmer would have to work harder to write code that: Runs as fast as Perl code Is represented by fewer lines of code Correctness Perl fully parses and pre- compiles script before execution. Efficiently eliminates the potential for runtime SYNTAX errors. Free to use Comes with source code.")
6
Hello, world! #!/usr/local/bin/perl # print “Hello, world \n”; interpreter path ‘#’ denotes a line commment Delimits a string Function which outputs arguments. Newline character Terminator character

7
Basic Program Flow No “main” function Statements executed from start to end of file. Execution continues until End of file is reached. exit(int) is called. Fatal error occurs.
 is called. Fatal error occurs..")
8
Variables Data of any type may be stored within three basic types of variables: Scalar List Associative array (hash table) Variables are always preceded by a “dereferencing symbol”. $ - Scalar variables @ - List variables % - Associative array variables

9
Variables Notice that we did NOT have to Declare the variable before using it Define the variable’s data type Allocate memory for new data values

10
Scalar variables References to variables always being with “$” in both assignments and accesses: For scalars: $x = 1; $x = “Hello World!”; $x = $y; For scalar arrays: $a[1] = 0; $a[1] = $b[1];
![Scalar variables References to variables always being with $ in both assignments and accesses: For scalars: $x = 1; $x = Hello World! ; $x = $y; For scalar arrays: $a[1] = 0; $a[1] = $b[1];](http://images.slideplayer.com/17/5334716/slides/slide_10.jpg "Scalar variables References to variables always being with $ in both assignments and accesses: For scalars: $x = 1; $x = Hello World! ; $x = $y; For scalar arrays: $a[1] = 0; $a[1] = $b[1];")
11
List variables Lists are prefaced by an “@” symbol: @count = (1, 2, 3, 4, 5); @count = (“apple”, “bat”, “cat”); @count2 = @count; A list is simply an array of scalar values. Integer indexes can be used to reference elements of a list. To print an element of an array, do: print $count[2];

12
Associative Array variables Associative array variables are denoted by the % dereferencing symbol. Associative array variables are simply hash tables containing scalar values Example: $fred{“a”} = “aaa”; $fred{“b”} = “bbb”; $fred{6} = “cc”; $fred{1} = 2; To do this in one step: %fred = (“a”, “aaa”, “b”, “bbb”, 6, “cc”, 1, 2);
;.")
13
Statements & Input/Output Statements Contains all the usual if, for, while, and more… Input/Output Any variable not starting with “$”, “@” or “%” is assumed to be a filehandle. There are several predefined filehandles, including STDIN, STDOUT and STDERR.

14
Subroutines We can reuse a segment of Perl code by placing it within a subroutine. The subroutine is defined using the sub keyword and a name. The subroutine body is defined by placing code statements within the {} code block symbols. sub MySubroutine { #Perl code goes here. }

15
Subroutine call To call a subroutine, prepend the name with the & symbol: &MySubroutine; Subroutine may be recursive (call themselves).
.")
16
Pattern Matching Perl enables to compare a regular expression pattern against a target string to test for a possible match. The outcome of the test is a boolean result (TRUE or FALSE). The basic syntax of a pattern match is $myScalar =~ / PATTERN / “Does $myScalar contain PATTERN ?”
. The basic syntax of a pattern match is $myScalar =~ / PATTERN / Does $myScalar contain PATTERN .")
17
Functions Perl provides a rich set of built-in functions to help you perform common tasks. Several categories of useful built-in function include Arithmetic functions (sqrt, sin, … ) List functions (push, chop, … ) String functions (length, substr, … ) Existance functions (defined, undef)
 List functions (push, chop, … ) String functions (length, substr, … ) Existance functions (defined, undef).")
18
Perl 5 Introduce new features: A new data type: the reference A new localization: the my keyword Tools to allow object oriented programming in Perl New shortcuts like “ qw ” and “ => ” An object oriented based liberary system focused around “Modules”

19
References A reference is a scalar value which “points to” any variable. Variable Value Reference

20
Creating References References to variables are created by using the backslash(\) operator. $name = “bio perl”; $reference = \$name; $array_reference = \@array_name; $hash_reference = \%hash_name; $subroutine_ref = \&sub_name;

21
Dereferencing a Reference Use an extra $ and @ for scalars and arrays, and -> for hashes. print “$$scalar_reference\n” “@$array_reference\n” “$hash_reference->{‘name’}\n”;

22
Variable Localization local keyword is used to limit the scope of a variable to within its enclosing brackets. Visible not only from within the enclosing bracket but in all subroutine called within those brackets $a = 1; sub mySub { local $a = 2; &mySub1($a); } sub mySub1 { print “a is $a\n”; } a is 2
; } sub mySub1 { print a is $a\n ; } a is 2.")
23
Variable Localization – cont’d my keyword hides the variable from the outside world completely. Totally hidden $a = 1; sub mySub { my $a = 2; &mySub1($a); } sub mySub1 { print “a is $a\n”; } a is 1
; } sub mySub1 { print a is $a\n ; } a is 1.")
24
Object Oriented Programming in Perl (1) Defining a class A class is simply a package with subroutines that function as methods. #!/usr/local/bin/perl package Cat; sub new { … } sub meow { … }

25
Object Oriented Programming in Perl (2) $new_object = new ClassName; $cat->meow(); Perl Object To initiates an object from a class, call the class “ new ” method. Using Method To use the methods of an object, use the “ -> ” operator.

26
Object Oriented Programming in Perl (3) Inheritance Declare a class array called @ISA. This array store the name and parent class(es) of the new species. package NorthAmericanCat; @NorthAmericanCat::ISA = (“Cat”); sub new { … }
 of the new species. package = ( Cat ); sub new { … }.")
27
Miscellaneous Constructs qw The “qw” keyword is used to bypass the quote and comma character in list array definitions. @name = (“Tom”, “Mary”, “Michael”); @name = qw(Tom Mary Michael);
;.")
28
Miscellaneous Constructs => The => operator is used to make hash definitions more readable. %client = {“name”,, “Michael”, “phone”, ”123-3456”, “email”, ”mich@nj.net”};mich@nj.net %client = {“name” => “Michael”, “phone” => ”123-3456”, “email” => “mich@nj.net”};mich@nj.net

29
Perl Modules A Perl module is a reusable package defined in a library file whose name is the same as the name of the package. Similar to C link library or C++ class package Foo; sub bar { print “Hello $_[0]\n”} sub blat { print “World $_[0]\n”: 1;

30
Names Each Perl module has a unique name. To minimize name space collision, Perl provides a hierarchical name space for modules. Components of a module name are separated by double colons (::). For example, Math::Complex Math::Approx String::BitCount String::Approx
. For example, Math::Complex Math::Approx String::BitCount String::Approx.")
31
Module files Each module is contained in a single file. Module files are stored in a subdirectory hierarchy that parallels the module name hierarchy. All module files have an extension of.pm. ModuleIs stored in ConfigConfig.pm Math::ComplexMath/Complex.pm String::ApproxString/Approx.pm

32
Module libraries The Perl interpreter has a list of directories in which it searhces for modules. Global arry @INC >perl –V @INC: /usr/local/lib/perl5/5.00503/sun4-solaris /usr/local/lib/perl5/5.00503 /usr/local/lib/perl5/site-perl/5.005/sun4-solaris /usr/local/lib/perl5/site-perl/5.005

33
Creating Modules To create a new Perl module:../development>h2xs –X –n Foo::Bar Writing Foo/Bar/Bar.pm Writing Foo/Bar/Makefile.PL Writing Foo/Bar/test.pl Writing Foo/Bar/Changes Writing Foo/Bar/MANIFEST../development>

34
Building Modules To build a Perl module: perl Makefile.PL make make test make install Create the makefile Create test directory blib and the installs the module in it. Run test.pl Install your module

35
Using Modules A module can be loaded by calling the use function. use Foo; bar( “a” ); blat( “b” ); Calls the eval function to process the code. The 1; causes eval to evaluate to TRUE.
; blat( b ); Calls the eval function to process the code. The 1; causes eval to evaluate to TRUE..")
36
End of Part I. Thank You…

37
Part II: XS(eXternal subroutine)extension Sen Zhang
extension Sen Zhang")
38
XS XS is an acronym for eXternal Subroutine. With XS, we can call C subroutines directly from Perl code, as if they were Perl subroutines.

39
Perl is not good at: very CPU-intensive things, like numerical integration. very memory-intensive things. Perl programs that create more than 10,000 hashes run slowly. system software, like device drivers. things that have already been written in other languages.

40
Usually… These things are done by other highly efficient system programming languages such as C\C++.

41
Can we call C subroutine from Perl? Solution is: Perl C API

42
When perl talks with C subroutine using perl C API two things must happen: control flow - control must pass from Perl to C (and back) C program execution Perl program execution data flow - data must pass from Perl to C (and back) C data representation Perl data representation
 C program execution Perl program execution data flow - data must pass from Perl to C (and back) C data representation Perl data representation")
43
In order to use perl C API What is Perl's internal data structures. How the Perl stack works, and how a C subroutine gets access to it. How C subroutines get linked into the Perl executable. Understand the data paths through the DynaLoader module that associate the name of a Perl subroutine with the entry point of a C subroutine

44
If you do code directly to the Perl C API You will find You keep writing the same little bits of code to move parameters on and off the Perl stack; to convert data from Perl's internal representation to C variables; to check for null pointers and other Bad Things. When you make a mistake, you don't get bad output: you crash the interpreter. It is difficult, error-prone, tedious, and repetitive.

45
Pain killer is XS

46
What is XS? Narrowly, XS is the name of the glue language More broadly, XS comprises a system of programs and facilities that work together : MakeMaker, Xsub glue routine, XS language itself, xsubpp, h2xs, DynaLoader.

47
MakeMaker -tool Perl's MakeMaker facility can be used to provide a Makefile to easily install your Perl modules and scripts.

48
MakeMaker, Xsub glue routine, XS language itself, xsubpp, h2xs, DynaLoader.

49
MakeMaker, Xsub glue routine, XS language itself, xsubpp, h2xs, DynaLoader.

50
Xsub The Perl interpreter calls a kind of glue routine as an xsub. Rather than drag the Perl C API into all our C code, we usually write glue routines. (We'll refer to an existing C subroutine as a target routine.)
.")
51
Xsub- control flow The glue routine converts the Perl parameters to C data values, and then calls the target routine, passing it the C data values as parameters on the processor stack. When the target routine returns, the glue routine creates a Perl data object to represent its return value, and pushes a pointer to that object onto the Perl stack. Finally, the glue routine returns control to the Perl interpreter.

52
Xsub-data flow Something has to convert between Perl and C data representations. The Perl interpreter doesn't, so the xsub has to. Typically, the xsub uses facilities in the Perl C API to get parameters from the Perl stack and convert them to C data values. To return a value, the xsub creates a Perl data object and leaves a pointer to it on the Perl stack.

53
MakeMaker, Xsub glue routine, XS language itself, xsubpp, h2xs, DynaLoader.

54
MakeMaker, Xsub glue routine, XS language itself, xsubpp, h2xs, DynaLoader.

55
XS - language Glue routines provide some structure for the data flow and control flow, but they are still hard to write. So we don't. Instead, we write XS code. XS is, more or less, a macro language. It allows us to declare target routines, and specify the correspondence between Perl and C data types. XS is a collection of macros, while Perl docs refer to XS as a language, it is a macro language.

56
MakeMaker, Xsub glue routine, XS language itself, xsubpp, h2xs, DynaLoader.

57
MakeMaker, Xsub glue routine, XS language itself, xsubpp, h2xs, DynaLoader.

58
Xsubpp-tool xsubpp is a XS language processor, xsubpp is the program that translates XS code to C code. xsubpp will compile XS code into C code by embedding the constructs necessary to let C functions manipulate Perl values and creates the glue necessary to let Perl access those functions. xsubpp expands XS macros into the bits of C code(xsub-glue routines) necessary to connect the Perl interpreter to your C- language subroutines. write XS code so that xsubpp will do the right thing.
 necessary to connect the Perl interpreter to your C- language subroutines. write XS code so that xsubpp will do the right thing..")
59
MakeMaker, Xsub glue routine, XS language itself, xsubpp, h2xs, DynaLoader.

60
MakeMaker, Xsub glue routine, XS language itself, xsubpp, h2xs, DynaLoader.

61
H2xs - tool h2xs was originally written to generate XS interfaces for existing C libraries. h2xs is a utility that reads a.h file and generates an outline for an XS interface to the C code.

62
MakeMaker, Xsub glue routine, XS language itself, xsubpp, h2xs, DynaLoader.

63
MakeMaker, Xsub glue routine, XS language itself, xsubpp, h2xs, DynaLoader.

64
DynaLoader-module In order for a C subroutine to become an xsub, three things must happen Loading:the subroutine has to be loaded into memory Linking:the Perl interpreter has to find its entry point Installation:the interpreter has to set the xsub pointer in a code reference to the entry point of the subroutine

65
DynaLoader. Fortunately, all this is done for us by a Perl module called the DynaLoader. When we write an XS module, our module inherits from DynaLoader. When our module loads, it makes a single call to the DynaLoader::bootstrap method. bootstrap locates our link libraries, loads them, finds our entry points, and makes the appropriate calls.

66
Perl module Development time Running time.c.h Complier, linker library h2xs XS code xsubpp Xsub(glue subrutine) Perl interprator Pure perl code Perl C API Output Input Some Manual change DynaLoader.
 Perl interprator Pure perl code Perl C API Output Input Some Manual change DynaLoader.")
67
An Example- Needleman-Wunsch(NW) Sequence alignment is an important problem in the bleeding-edge field of genomics. Sequence alignment is a combinatorial problem, and naive algorithms run in exponential time. The Needleman-Wunsch algorithm runs in (more or less) O(n^3),O(n^3) Dynamic programming algorithm for global optimal sequence alignment.
 O(n^3),O(n^3) Dynamic programming algorithm for global optimal sequence alignment..")
68
Algorithm

69
Score matrix

70
Complexity analysis The O(n^3) step in the NW algorithm is filling in the score matrix; everything else runs in linear time. We want tolinear time use the C implementation to fill in the score matrix, use the Perl implementation for everything else, and use XS to call from one to the other.

71
Our approach Implement the algorithm as a straight Perl module Analyze (or benchmark) the code for performance Reimplement performance-critical methods (score matrix filling) in C Write XS to connect the C routines to the Perl module
 the code for performance Reimplement performance-critical methods (score matrix filling) in C Write XS to connect the C routines to the Perl module")
72
Performance comparison a straight Perl implementation of the NW algorithm aligns 2 200-character sequences in 300 seconds.straight Perl implementation XS version runs the benchmark 200x200 alignment in 3 seconds. XS version is about 100 times faster than the Perl implementation.

73
Bio::Tools::pSW - pairwise Smith Waterman object Bioperl project has pSW implementation. pSW is an Alignment Factory. It builds pairwise alignments using the smith waterman algorithm. The alignment algorithm is implemented in C and added in using an XS extension. The Smith-Waterman algorithm needs O(n^2) time to find the highest scoring cell in the matrix.
 time to find the highest scoring cell in the matrix..")
74
The end of Part II Thanks

75
Bioperl Introduction Hairong Zhao

76
What’s Bioperl? Bioperl is not a new language It is a collection of Perl modules that facilitate the development of Perl scripts for bio-informatics applications.

77
Perls script Perl Interpreter Perl Modules Bioperl Modules output input Bioperl and Perl

78
Why Bioperl for Bio-informatics? Perl is good at file manipulation and text processing, which make up a large part of the routine tasks in bio-informatics. Perl language, documentation and many Perl packages are freely available. Perl is easy to get started in, to write small and medium-sized programs.

79
Bioperl Project It is an international association of developers of open source Perl tools for bioinformatics, genomics and life science research Started in 1995 by a group of scientists tired of rewriting BLAST and sequence parsers for various formats Now there are 45 registered developers, 10-15 main developers, 5 core coordinate developers Project website:http://bioperl.org Project FTP server: bioperl.org

80
How many people use Bioperl? Bioperl has been used worldwide in both small academic labs through to enterprise level computing in large pharmaceutical companies since 1998 Bioperl Usage Survey http://www.bioperl.org/survey.html

81
The current status of Bioperl The latest mature and stable version 1.0 was released in March 2002. This new version contains 832 files. The test suite contains 93 scripts which collectively perform 3042 functionality tests. This new version is "feature complete" for sequence handling, the most common task in bioinformatics, it adds some new features and improve some existing features

82
The future of Bioperl It is far from mature: Except sequence handling, all other modules are not complete. The portability is not very good, not all modules will work with on all platforms.

83
Bioperl resources www.bioperl.org http://www.bioperl.org/Core/bptutorial.html Example code, in the scripts/ and examples/ directories. Online course written at the Pasteur Institute. See: http://www.pasteur.fr/recherche/unites/sis/f ormation/bioperl. http://www.pasteur.fr/recherche/unites/sis/f ormation/bioperl

84
Biopython, biojava Similar goals implemented in different language Most effort to date has been to port Bioperl functionality to Biopython and Biojava, so the differences are fairly peripheral In the future, some bio-informatics tasks may prove to be more effectively implemented in java or python, interoperability between them is necessary CORBA is one such framework for interlanguage support, and the Biocorba project is currently implementing a CORBA interface for bioperl

85
Bioperl-Object Oriented The Bioperl takes advantages of the OO design to create a consistent, well documented, object model for interacting with biological data in the life sciences. Bioperl Name space The Bioperl package installs everything in the Bio:: namespace.

86
Bioperl Objects Sequence handling objects Sequence objects Alignment objects Location objects Other Objects: 3D structure objects, tree objects and phylogenetic trees, map objects, bibliographic objects and graphics objects

87
Sequence handling Typical sequence handling tasks: Access the sequence Format the sequence Sequence alignment and comparison Search for similar sequences Pairwise comparisons Multiple alignment

88
Sequence Objects Sequence objects: Seq, RichSeq, SeqWithQuality, PrimarySeq, LocatableSeq, LiveSeq, LargeSeq, SeqI Seq is the central sequence object in bioperl, you can use it to describe a DNA, RNA or protein sequence. Most common sequence manipulations can be performed with Seq.

89
Sequence Annotation Bio::SeqFeature Sequence object can have multiple sequence feature (SeqFeature) objects - eg Gene, Exon, Promoter objects - associated with it. Bio::Annotation A Seq object can also have an Annotation object (used to store database links, literature references and comments) associated with it
 associated with it.")
90
Sequence Input/Output The Bio::SeqIO system was designed to make getting and storing sequences to and from the myriad of formats as easy as possible.

91
Diagram of Objects and Interfaces for Sequence Analysis

92
Accessing sequence data Bioperl supports accessing remote databases as well as local databases. Bioperl currently supports sequence data retrieval from the genbank, genpept, RefSeq, swissprot, and EMBL databases

93
Format the sequences SeqIO object can read a stream of sequences in one format: Fasta, EMBL, GenBank, Swissprot, PIR, GCG, SCF, phd/phred, Ace, or raw (plain sequence), then write to another file in another format use Bio::SeqIO; $in = Bio::SeqIO->new('-file' => "inputfilename", '-format' => 'Fasta'); $out = Bio::SeqIO->new('-file' => ">outputfilename", '-format' => 'EMBL'); while ( my $seq = $in->next_seq() ) {$out->write_seq($seq); }
, then write to another file in another format use Bio::SeqIO; $in = Bio::SeqIO->new( -file => inputfilename , -format => Fasta ); $out = Bio::SeqIO->new( -file => >outputfilename , -format => EMBL ); while ( my $seq = $in->next_seq() ) {$out->write_seq($seq); }")
94
Manipulating sequence data $seqobj->display_id(); # the human read-able id of the sequence $seqobj->subseq(5,10); # part of the sequence as a string $seqobj->desc() # a description of the sequence $seqobj->trunc(5,10) # truncation from 5 to 10 as new object $seqobj->revcom # reverse complements sequence $seqobj->translate # translation of the sequence …
; # the human read-able id of the sequence $seqobj->subseq(5,10); # part of the sequence as a string $seqobj->desc() # a description of the sequence $seqobj->trunc(5,10) # truncation from 5 to 10 as new object $seqobj->revcom # reverse complements sequence $seqobj->translate # translation of the sequence …")
95
Alignment Searching for ``similar'' sequences, Bioperl can run BLAST locally or remotely, and then parse the result. Aligning 2 sequences with Smith-Waterman (pSW) or blast The SW algorithm itself is implemented in C and incorporated into bioperl using an XS extension. Aligning multiple sequences (Clustalw.pm, TCoffee.pm) bioperl offers a perl interface to the bioinformatics- standard clustalw and tcoffee programs. Bioperl does not currently provide a perl interface for running HMMER. However, bioperl does provide a HMMER report parser.
 or blast The SW algorithm itself is implemented in C and incorporated into bioperl using an XS extension. Aligning multiple sequences (Clustalw.pm, TCoffee.pm) bioperl offers a perl interface to the bioinformatics- standard clustalw and tcoffee programs. Bioperl does not currently provide a perl interface for running HMMER. However, bioperl does provide a HMMER report parser..")
96
Alignment Objects Early versions used UnivAln, SimpleAlign Ver. 1.0 only support SimpleAlign. It allows the user to: convert between alignment formats extracting specific regions of the alignment generating consensus sequences. …

97
Sequence handling objects Sequence objects Alignment objects Location objects

98
Location Objects Bio::Locations: a collection of rather complicated objects A Location object is designed to be associated with a Sequence Feature object to indicate where on a larger structure (eg a chromosome or contig) the feature can be found.
 the feature can be found.")
99
Conclusion Bioperl is Powerful Easy Waiting for you (biologist) to use
 to use")
100
Scripts Examples by Using Bioperl Tiequan zhang

101
SimpleAlign module Description: It handles multiple alignments of sequences Lightweight display/formatting and minimal manipulation

102
Method: new Usage : my $aln = new Bio::SimpleAlign(); Function : Creates a new simple align object Returns : Bio::SimpleAlign Args : -source => string representing the source program where this alignment came from each_seq Usage : foreach $seq ( $align->each_seq() ) Function : Gets an array of Seq objects from the alignment Returns : an array length() Usage : $len = $ali->length() Function : Returns the maximum length of the alignment. To be sure the alignment is a block, use is_flush

103
consensus_string Usage : $str = $ali->consensus_string($threshold_percent) Function : Makes a strict consensus Args : Optional treshold ranging from 0 to 100. The consensus residue has to appear at least threshold % of the sequences at a given location, otherwise a '?' character will be placed at that location. (Default value = 0%) is_flush Usage : if( $ali->is_flush() ) Function : Tells you whether the alignment is flush, ie all of the same length Returns : 1 or 0 percentage_identity Usage : $id = $align->percentage_identity Function: The function calculates the average percentage identity Returns : The average percentage identity no_sequences Usage : $depth = $ali->no_sequences Function : number of sequence in the sequence alignment Returns : integer
 is_flush Usage : if( $ali->is_flush() ) Function : Tells you whether the alignment is flush, ie all of the same length Returns : 1 or 0 percentage_identity Usage : $id = $align->percentage_identity Function: The function calculates the average percentage identity Returns : The average percentage identity no_sequences Usage : $depth = $ali->no_sequences Function : number of sequence in the sequence alignment Returns : integer.")
104
testaln.pfam 1433_LYCES/9-246 REENVYMAKLADRAESDEEMVEFMEKVSNSLGS.EELTVEERNLLSVAYKNVIGARRAS$ 1434_LYCES/6-243 REENVYLAKLAEQAERYEEMIEFMEKVAKTADV.EELTVEERNLLSVAYKNVIGARRAS$ 143R_ARATH/7-245 RDQYVYMAKLAEQAERYEEMVQFMEQLVTGATPAEELTVEERNLLSVAYKNVIGSLRAA$ 143B_VICFA/7-242 RENFVYIAKLAEQAERYEEMVDSMKNVANLDV...ELTIEERNLLSVGYKNVIGARRAS$ 143E_HUMAN/4-239 REDLVYQAKLAEQAERYDEMVESMKKVAGMDV...ELTVEERNLLSVAYKNVIGARRAS$ BMH1_YEAST/4-240 REDSVYLAKLAEQAERYEEMVENMKTVASSGQ...ELSVEERNLLSVAYKNVIGARRAS$ RA24_SCHPO/6-241 REDAVYLAKLAEQAERYEGMVENMKSVASTDQ...ELTVEERNLLSVAYKNVIGARRAS$ RA25_SCHPO/5-240 RENSVYLAKLAEQAERYEEMVENMKKVACSND...KLSVEERNLLSVAYKNIIGARRAS$ 1431_ENTHI/4-239 REDCVYTAKLAEQSERYDEMVQCMKQVAEMEA...ELSIEERNLLSVAYKNVIGAKRAS$

105
Script: use Bio::AlignIO $str = Bio::AlignIO->new('-file' => 'testaln.pfam'); $aln = $str->next_aln(); print $aln->length, "\n"; print $aln->no_residues, "\n"; print $aln->is_flush, "\n"; print $aln->no_sequences, "\n"; print $aln->percentage_identity, "\n"; print $aln->consensus_string(50), "\n"; $pos = $aln->column_from_residue_number('1433_LYCES', 14); # = 6; foreach $seq ($aln->each_seq) { $res = $seq->subseq($pos, $pos); $count{$res}++; } foreach $res (keys %count) { printf "Res: %s Count: %2d\n", $res, $count{$res}; }
; $aln = $str->next_aln(); print $aln->length, \n ; print $aln->no_residues, \n ; print $aln->is_flush, \n ; print $aln->no_sequences, \n ; print $aln->percentage_identity, \n ; print $aln->consensus_string(50), \n ; $pos = $aln->column_from_residue_number( 1433_LYCES , 14); # = 6; foreach $seq ($aln->each_seq) { $res = $seq->subseq($pos, $pos); $count{$res}++; } foreach $res (keys %count) { printf Res: %s Count: %2d\n , $res, $count{$res}; }")
106
Result: argerich-54 bio>: perl align.pl 242 103 1 16 66.9052451661147 RE??VY?AKLAEQAERYEEMV??MK?VAE??????ELSVEERNLLSVAYKNVIGARRAS WRIISSIEQKEE??G?N?????LIKEYR?KIE?EL??IC?DVL?LLD??LIP?A?????ESKV FYLKMKGDYYRYLAEFA?G??RKE?AD?SL?AYK?A?DIA?AEL?PTHPIRLGLALNFS VFYYEILNSPD?AC?LAKQAFDEAIAELDTL?EESYKDSTLIMQLLRDNLTLWTSD?? ??? Res: Q Count: 5 Res: Y Count: 10 Res:. Count: 1 argerich-55 bio>:

107
SwissProt,Seq and SeqIO modules Description: SwissProt is a curated database of proteins managed by the Swiss Bioinformatics Institute. This is in contrast to EMBL/GenBank/DDBJ Which are archives of protein information. It allows the dynamic retrieval of Sequence objects (Bio::Seq)
.")
108
SeqIO can be used to convert different formats: 1. Fasta FASTA format 2. EMBL EMBL format 3. GenBank GenBank format 4. swiss Swissprot format 5. SCF SCF tracefile format 6. PIR Protein Information Resource format 7. GCG GCG format 8. raw Raw format 9. ace ACeDB sequence format

109
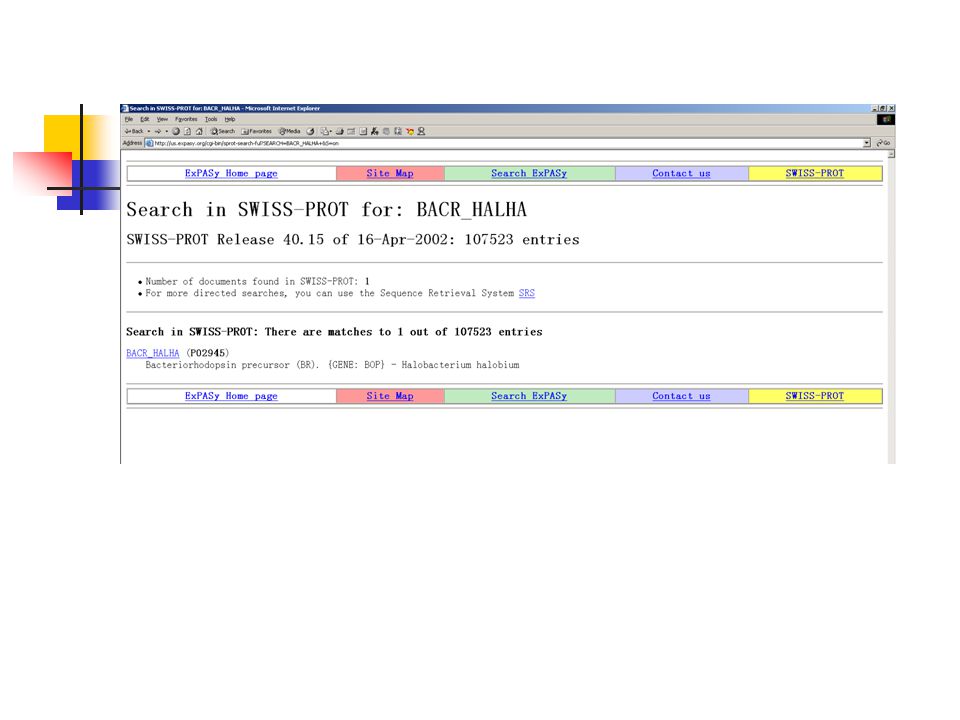
Objective: loading a sequence from a remote server Create a sequence object for the BACR_HALHA SwissProt entry Print its Accession number and description Display the sequence in FASTA format

112
Scripts: #!/usr/bin/perl use strict; use Bio::DB::SwissProt; use Bio::Seq; use Bio::SeqIO; my $database = new Bio::DB::SwissProt; my $seq = $database->get_Seq_by_id('BACR_HALHA'); print "Seq: ", $seq->accession_number(), " -- ", $seq->desc(), "\n\n"; my $out = Bio::SeqIO->newFh ( -fh => \*STDOUT, -format => 'fasta'); print $out $seq;
; print Seq: , $seq->accession_number(), -- , $seq->desc(), \n\n ; my $out = Bio::SeqIO->newFh ( -fh => \*STDOUT, -format => fasta ); print $out $seq;")
113
Result: argerich-47 bio>: perl protein.pl Seq: P02945 -- BACTERIORHODOPSIN PRECURSOR (BR). >BACR_HALHA BACTERIORHODOPSIN PRECURSOR (BR). MLELLPTAVEGVSQAQITGRPEWIWLALGTALMGLGTLYFLVKGMGVSDPDAKKFYAITT LVPAIAFTMYLSMLLGYGLTMVPFGGEQNPIYWARYADWLFTTPLLLLDLALLVDADQGT ILALVGADGIMIGTGLVGALTKVYSYRFVWWAISTAAMLYILYVLFFGFTSKAESMRPEV ASTFKVLRNVTVVLWSAYPVVWLIGSEGAGIVPLNIETLLFMVLDVSAKVGFGLILLRSR AIFGEAEAPEPSAGDGAAATSD argerich-48 bio>:
. MLELLPTAVEGVSQAQITGRPEWIWLALGTALMGLGTLYFLVKGMGVSDPDAKKFYAITT LVPAIAFTMYLSMLLGYGLTMVPFGGEQNPIYWARYADWLFTTPLLLLDLALLVDADQGT ILALVGADGIMIGTGLVGALTKVYSYRFVWWAISTAAMLYILYVLFFGFTSKAESMRPEV ASTFKVLRNVTVVLWSAYPVVWLIGSEGAGIVPLNIETLLFMVLDVSAKVGFGLILLRSR AIFGEAEAPEPSAGDGAAATSD argerich-48 bio>:.")
114
Summary Perl language and modules Perl XS Bioperl Example scripts

115
References: [1] L. Wall and R. Schwarz. Programming Perl. O’Reilly & Associates, Inc, 1991. [2] Web Developer’s Virtual Library. http://www.wdvl.com/Authoring/Languages/Perl/5/ http://www.wdvl.com/Authoring/Languages/Perl/5/ [3] O’Reily Perl.com. http://www.perl.com/ [4] http://archive.ncsa.uiuc.edu/General/Training/PerlIntro/ [5] http://www.vis.ethz.ch/manuals/Perl/intro.html [6] http://www.fukada.com/selena/tutorials/perl5/index.html [7] http://world.std.com/~swmcd/steven/perl/module_mechanics.html [8] http://www.sdsc.edu/~moreland/courses/IntroPerl/ [9] www.bioperl.org/Core/POD/Bio/SeqIO.htmlwww.bioperl.org/Core/POD/Bio/SeqIO.html [10] http://docs.bioperl.org/releases/bioperl-1.0/Bio/SimpleAlign.htmlhttp://docs.bioperl.org/releases/bioperl-1.0/Bio/SimpleAlign.html [11] www.pasteur.fr/recherche/unites/sis/formation/bioperl/index.htmlwww.pasteur.fr/recherche/unites/sis/formation/bioperl/index.html
![References: [1] L. Wall and R. Schwarz. Programming Perl.](http://images.slideplayer.com/17/5334716/slides/slide_115.jpg "O’Reilly & Associates, Inc, [2] Web Developer’s Virtual Library. [3] O’Reily Perl.com. [4] [5] [6] [7] [8] [9] [10] [11]")
116
References: [12] www.bioinformatics.comwww.bioinformatics.com [13] ] Bioperl: Standard Perl Modules for Bioinformatics by Stephen A Chervitz, Georg Fuellen, Chris Dagdigian, Steven E Brenner, Ewan Birney and Ian Korf Objects in Bioinformatics '98Objects in Bioinformatics '98 [15] http://cvs.open-bio.org/cgi-bin/viewcvs/viewcvs.cgi/bioperl- papers/bioperldesignhttp://cvs.open-bio.org/cgi-bin/viewcvs/viewcvs.cgi/bioperl- papers/bioperldesign [16] http://www.cpan.orghttp://www.cpan.org [17] http://www.maths.tcd.ie/~lily/pres2/sld008.htmhttp://www.maths.tcd.ie/~lily/pres2/sld008.htm [18] http://www.sbc.su.se/~per/molbioinfo2001/dynprog/dynamic.htmlhttp://www.sbc.su.se/~per/molbioinfo2001/dynprog/dynamic.html [19] http://world.std.com/~swmcd/steven/perl/pm/xs/intro/index.htmlhttp://world.std.com/~swmcd/steven/perl/pm/xs/intro/index.html
![References: [12] [13] ] Bioperl: Standard Perl Modules for Bioinformatics by Stephen A Chervitz, Georg Fuellen, Chris Dagdigian, Steven E Brenner, Ewan Birney and Ian Korf Objects in Bioinformatics 98Objects in Bioinformatics 98 [15] papers/bioperldesignhttp://cvs.open-bio.org/cgi-bin/viewcvs/viewcvs.cgi/bioperl- papers/bioperldesign [16] [17] [18] [19]](http://images.slideplayer.com/17/5334716/slides/slide_116.jpg "References: [12] [13] ] Bioperl: Standard Perl Modules for Bioinformatics by Stephen A Chervitz, Georg Fuellen, Chris Dagdigian, Steven E Brenner, Ewan Birney and Ian Korf Objects in Bioinformatics 98Objects in Bioinformatics 98 [15] papers/bioperldesignhttp://cvs.open-bio.org/cgi-bin/viewcvs/viewcvs.cgi/bioperl- papers/bioperldesign [16] [17] [18] [19]")
Similar presentations
>")



 methods and.>")







 Tier architecture PHP script Remote services Web Server (Apache, IIS) Browser (IE, FireFox, Opera) Desktop (PC or MAC)>")

 $seq =~ tr/ACGT/TGCA/; $seq = reverse $seq; return $seq; } my $revSeq = reverseComplement(GCAGTG);>")

 provides new flexible ways for defining.>")


