Download presentation
Presentation is loading. Please wait.

1
Building Phylogenies Distance-Based Methods

2
Methods Distance-based Parsimony Maximum likelihood

3
Distance Matrices a0 b60 c730 d141090 abcd a b c d 123450678 Distance matrix is additive if there is a tree that fits it exactly

4
Ultrametric Matrices a0 b20 c660 d1010100 abcd a b c d 123450 Additive + molecular clock assumption

5
Methods Fitch - Margoliash UPGMA Neighbor-joining Many others

6
Least squares trees Minimize over all trees Choice of weights w ij : –Uniform: w ij 1 –Fitch-Margoliash: w ij 1/D ij 2 –Others...

7
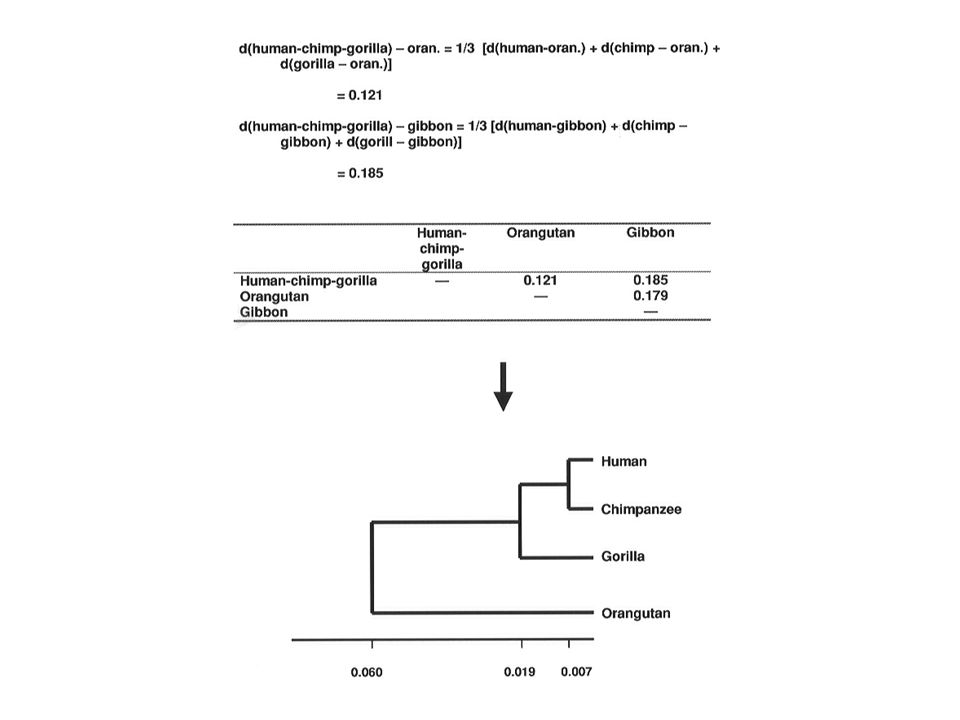
Sarich's (1969) immunological distances
 immunological distances")
8
Least squares tree for Sarich’s data

9
Clustering Methods E.g., UPGMA and Neighbor-Joining A cluster is a set of taxa Interspecies distances translate into intercluster distances Clusters are repeatedly merged –“Closest” clusters merged first –Distances are recomputed after merging

10
UPGMA Unweighted pair group method using arithmetic averages The distance between clusters C i and C j is After merging C i and C j to create cluster C k define distance from k to every other cluster r as

11
UPGMA: Initialization 1.Assign each sequence i to its own cluster C i 2.Define one leaf (tip) of tree for each sequence and place it at height 0
 of tree for each sequence and place it at height 0")
12
UPGMA: Iteration 1.Choose the two clusters i and j with smallest D ij 2.Create a new cluster k, where C k = C i C j 3.Compute D kr for all r. 4.Define a new node k with children i and j, and place it at height D ij /2. 5.Add k to the current clusters and delete i and j Let i and j be the remaining clusters. Place root at height D ij /2 Repeat until only two clusters remain:

13
UPGMA Example

17
UPGMA tree for Sarich’s data

18
A pitfall of UPGMA The algorithm produces an ultrametric tree: the distance from the root to any leaf is the same UPGMA assumes a constant molecular clock: all species accumulate mutations (evolve) at the same rate.
 at the same rate.")
19
UPGMA fails when molecular clock assumption doesn’t hold

20
Neighbor Joining Saitou and Nei, Molecular Biology and Evolution 4 (1987) Idea: Find a pair of leaves that are close to each other but far from other leaves –Implicitly finds a pair of neighboring leaves Advantages: –Works well for additive and other nonadditive matrices –Does not have the molecular clock assumption
 Idea: Find a pair of leaves that are close to each other but far from other leaves –Implicitly finds a pair of neighboring leaves Advantages: –Works well for additive and other nonadditive matrices –Does not have the molecular clock assumption")
21
Long branches must be handled carefully! 0.1 0.4 and are closer to each other than to or . Obvious approach produces incorrect clusters!

22
Compensating for long edges Introduce “correction terms” “Corrected” distances: Distances are reduced for pairs that are far away from all other species: They may be close to each other. Average dist. to other taxa

23
Neighbor-joining 1.Choose i, j such that D ij u i u j is minimum 2.Define a new leaf k whose distances to i and j are 3.Compute the distance from k to every other leaf r 4.Delete i and j Repeat the following until only two leaves remain: Connect the 2 remaining leaves by a branch of length D ij

24
NJ tree for Sarich’s data

25
Computing distance matrices Based on sequence alignment Various possibilities: –Distance = average number of differences –Try different PAM matrices; distance = index of matrix that gives highest score –Feng and Doolitle: Based on alignment scores – roughly ratio to max possible score (see text) Read, e.g., PHYLIP documentation: http://evolution.genetics.washingt on.edu/phylip/general.html http://evolution.genetics.washingt on.edu/phylip/general.html
 Read, e.g., PHYLIP documentation: on.edu/phylip/general.html on.edu/phylip/general.html")
26
Distance correction The amount of evolutionary change is not linearly related to time Over a long period of time, a series of substitutions may bring us back to where we started Percentage difference may underestimate evolutionary time

27
Jukes-Cantor Model

28
Correcting for multiple substitutions in the JC model

29
Many other models!

Similar presentations




 or Evolutionary tree represents the evolutionary relationships among a set of organisms or groups of.>")













 Modified by Benny Chor, from slides by Shlomo Moran and Ydo Wexler (IIT)>")


