Download presentation
Presentation is loading. Please wait.

1
Today’s Agenda John M.’s presentation (15-25 min) Phylogenetic Trees – Overview – Construction – Algorithms – Etc.
 Phylogenetic Trees – Overview – Construction – Algorithms – Etc.")
2
Phylogenetic Trees The means by which biologists portray – the history of life over millions of years and – the branching events that gave rise to biological diversity

3
Phylogenetic Trees Phylogenetic trees of different alleles of a particular gene are the byproduct of 1. many rounds of mutation, 2. drift and 3. selection resulting in nearly unique sequences at that allele for individuals within species.

4
Phylogenetic Trees

5
Questions You Should Be Asking: What are Alleles? What exactly is mutation; what causes it? What is drift? What is selection?

6
Alleles & Genes Remember a gene is a segment of DNA that ultimately encodes a protein – A protein that performs an important biological function. – Or leads to some other important trait Very similar organisms have the same genes – For example, we all have the gene for hemoglobin – However, a slight change in that gene might result in sickle cell anemia – Another slight change might not cause any effect – A severe change might lead to a complete failure to create hemoglobin (i.e., death) Different versions of a gene are called alleles.
 Different versions of a gene are called alleles..")
7
Natural Selection A prime objective for all species is to reproduce and survive, When species do this they tend to produce more offspring than the environment can support. The lack of resources to nourish these individuals places pressure on the size of the species population, and the lack of resources means increased competition and as a consequence, some organisms will not survive.

8
Natural Selection The organisms who die as a consequence of this competition were not totally random, Darwin found that those organisms more suited to their environment were more likely to survive. Those organisms who are better suited to their environment exhibit desirable characteristics, which is a consequence of their genome being more suitable to begin with.

9
Natural Selection As a particular species spreads over a large geographic area, genetic branches arise. Different areas (geographic regions) provide different selection criteria A sub-population of a species might find itself isolated in tropical environment While another sub-population get isolated in a desert environment
 provide different selection criteria A sub-population of a species might find itself isolated in tropical environment While another sub-population get isolated in a desert environment.")
15
Mutation A mutation or polymorphism is a change in the DNA "letters" of a gene or an alteration in the chromosomes. Most DNA variation is neutral (not beneficial or harmful), But harmful sequence changes sometimes do occur. Changes within genes can result in proteins that don't work normally or don't work at all. Some of these changes can contribute to disease or affect how someone responds to a medicine.
, But harmful sequence changes sometimes do occur. Changes within genes can result in proteins that don t work normally or don t work at all. Some of these changes can contribute to disease or affect how someone responds to a medicine..")
16
Mutation Mutations 1. may be passed down from parent to child (in the sperm or egg cells), 2. may occur around the time of conception or 3. may be acquired during a person's lifetime. Can arise spontaneously during normal cell functions – when a cell divides, or – in response to environmental factors such as toxins, radiation, hormones, and even diet.

17
Mutation Nature provides us with a system of finely tuned repair enzymes that find and fix most DNA errors. But as our bodies change in response to age, illness and other factors, our repair systems may become less efficient. Uncorrected mutations can accumulate, resulting in nasty stuff.

18
Genetic Drift Allele frequencies can change due to chance alone. Alleles that form the next generation's gene pool are a sample of the alleles from the current generation. When sampled from a population, the frequency of alleles differs slightly due to chance alone. A small percentage of alleles may continually change frequency in a single direction for several generations – just as flipping a fair coin may, on occasion, result in a string of heads or tails.

19
Next Generation Genetic Drift Next Generation Parent Population

20
Genetic Drift Sharp drops in population size can change allele frequencies substantially. When a population crashes, the alleles in the surviving sample may not be representative of the pre-crash gene pool. This change in the gene pool is called the founder effect, because small populations of organisms that invade a new territory (founders) are subject to this. Many biologists feel the genetic changes brought about by founder effects may contribute to isolated populations developing reproductive isolation from their parent populations.
 are subject to this. Many biologists feel the genetic changes brought about by founder effects may contribute to isolated populations developing reproductive isolation from their parent populations..")
21
Genetic Drift The founders effect Invaders Large Population Small subset Survives

22
Genetic Drift & Fitness Large populations are often divided into smaller subpopulations. – Drift causes allele frequency differences between subpopulations If a subpopulation is small enough, the population could even drift through fitness valleys in the adaptive landscape. Then, the subpopulation could climb a larger fitness hill.

23
Genetic Drift & Fitness

24
Both natural selection and genetic drift decrease genetic variation. If they were the only mechanisms of evolution, populations would eventually become homogeneous and further evolution would be impossible. There are, however, mechanisms that replace variation depleted by selection and drift. Thank God for mutation and environmental diversity.

25
Trees and Distance http://babbage.clarku.edu/~djoyce/java/Phyltr ee/intro.html http://babbage.clarku.edu/~djoyce/java/Phyltr ee/intro.html

26
Trees and Distance

27
Reconstructing Phylogenetic Trees There are ten extant species (species currently living) – named from 1 through 10. The lines above the extant species represent the same species, just in the past.

28
Reconstructing Phylogenetic Trees When two lines converge to a point, that should be interpreted as the point when the two species diverged from a common ancestral species the point being the common ancestral species.

29
Reconstructing Phylogenetic Trees horizontal dimension doesn't mean anything! It is completely arbitrary whether a branch of the tree is placed to the left or to the right

30
Reconstructing Phylogenetic Trees vertical dimension corresponds to time. Although its imprecise, the difference between two species can be used to estimate when they diverged.

31
Reconstructing Phylogenetic Trees A tree isn't always the best model. Here are some times when it isn't best. For individuals within a species. The genetic material of an individual doesn't derive from a single earlier existing individual. – Animals and plants that multiply by sexual reproduction receive half their genetic material from each of two parents, so a tree like this is inappropriate.

32
Reconstructing Phylogenetic Trees Here are some other examples For closely related species. Individuals do occasionally mate between closely related species, and their progeny survive to contribute to the gene pool of one or both of the parent species. Hybrid species. In the plant world it occasionally happens that a new tetraploid species arises from two diploid species. The two parent species need to be somewhat related for this to happen.

33
Reconstructing Phylogenetic Trees Here is one last example: Distant interaction. There are a couple of ways that genetic material from one species can find its way into unrelated species. – Sometimes a bacterium of one species can ingest the genetic material of a bacterium of another species and incorporate part of it into its own genetic material. – Sometimes viruses can inadvertently transport genetic material from one species to another. In spite of these exceptions, a tree model is usually a pretty good model to show the relations among species.

34
Mutation Rates & Vertical Dimension Differences among species are the key to reconstructing the phylogenetic tree. Species differ in the characteristics, also called characters. The characters may be observable and measurable properties of the individuals. For instance, among mammals, the numbers of the different kinds of teeth that the individuals of the species have has been a successful character to classify mammals. This character has been especially important among extinct species since fossilized teeth are commonly found.

35
Mutation Rates & Vertical Dimension Any characters can be used to classify species and reconstruct a phylogenetic tree of species, – but some are more useful than others. If a species depends on a character for its continued survival, that character will not change as any mutations of it will be eliminated. Call such characters essential. And most visible characters are essential for the species. This means that if we choose essential characters, any differences should count as very significant.

36
Mutation Rates & Vertical Dimension There are, however, some difficulties with considering essential characters. If one species evolves by changing an essential characteristic, whatever ecological forces supported that change may also apply to other species, and that could lead to parallel evolution. Thus, differences or similarities in essential characters are very relevant to the reconstruction of the general shape of the phylogenetic tree, but they really can't be used to determine the relative lengths of the lines within the tree. Some species have been stable for millions of years. Others evolve very fast.

37
Mutation Rates & Vertical Dimension Irrelevant mutations. We could, on the other hand, consider nonessential characters. Changes in nonessential characters are effected by mutations, mutations that we can call irrelevant. The rate of change of irrelevant mutations should be fairly uniform among species, especially among species that are fairly closely related.

38
Mutation Rates & Vertical Dimension Much of the genome sequence of an organism is irrelevant. For example, there are 64 (4 3 ) different codons for 20 amino acids. Some amino acids are coded by up to four different codons. For these multiply coded amino acids, typically the third nucleotide can take any of the four possible values. In other words, a mutation in this third nucleotide is irrelevant. The DNA can mutate at this site and the resulting protein doesn't change.
 different codons for 20 amino acids. Some amino acids are coded by up to four different codons. For these multiply coded amino acids, typically the third nucleotide can take any of the four possible values. In other words, a mutation in this third nucleotide is irrelevant. The DNA can mutate at this site and the resulting protein doesn t change..")
39
Mutation Rates & Vertical Dimension By concentrating on irrelvant mutations, not only can the shape of the phylogenetic tree be reconstructed, but the relative lengths of the lines within the phylogenic tree can also be estimated.

40
Mutations as a measure of time Let's concentrate on one character to begin with. Our first questions are: – What is the probability p(t) that the character has some value at the beginning of a time interval of length t as it does at the end? – What is the probability q(t) that the character has one value at the beginning of a time interval of length t but a different value at the end of the interval?
 that the character has some value at the beginning of a time interval of length t as it does at the end. – What is the probability q(t) that the character has one value at the beginning of a time interval of length t but a different value at the end of the interval .")
41
Mutations as a measure of time Suppose that there are m different possible alternate values, and suppose that the mutation rate is r mutations per unit time interval. Some statistical analysis (which we'll skip) gives us the answers to these questions.
 gives us the answers to these questions..")
42
Mutations as a measure of time Note that initially, when t = 0, p(0) is 1, while q(0) is 0 since there are no mutations in no time. Also, as t approaches infinity, p(t) and q(t) both approach 1/m, which means that in the long run, each of the m alternative values are equally probable.
 and q(t) both approach 1/m, which means that in the long run, each of the m alternative values are equally probable..")
43
Mutations as a measure of time Now let's assume that there are n different characters, not just one. Then E(t), the expected number of characters that are not the same at the end of a time interval of length t as they were at the beginning, is n(m –1) q(t), that is,
, the expected number of characters that are not the same at the end of a time interval of length t as they were at the beginning, is n(m –1) q(t), that is,.")
44
Mutations as a measure of time Here's the graph of that function when there are m = 4 alternate values for each character, there are n = 40 characters, and the mutation rate is r = 0.1.

45
Mutations as a measure of time Time t is shown on the horizontal axis, while the vertical axis gives y, the expected number of character differences. Note that when t gets large, the expected number of character differences approaches 30.

46
Mutations as a measure of time We can take the inverse function of y = E(t), that is, turn this graph around, to give us an estimate for time t in terms of the observed number of character differences. Let g denote the inverse function. The base of the logarithm function here is e.

47
Mutations as a measure of time The graph of t = g(y) is shown to the right with the same parameter values m = 4, n = 40, and r = 0.1. Note that as the number of expected differences approaches 30, the corresponding time approaches infinity.

48
Mutations as a measure of time The observed number of differences may be near the expected number, but it's usually more or less. So the observed number of differences could easily be greater than 30.

49
Mutations as a measure of time Should that happen, the best conclusion to make is that the time is very great, but can't be estimated. It would be prudent not to estimate the time when the number of differences is slightly less than 30, too

50
Reconstruction How do you reconstruct the phylogenic tree when all you know are characters of extant species? When there are only a few species, only a few characters, and the number of mutations is small but not too small, then common sense and a little bit of logic does a pretty good job, at least for deciding on the shape of the tree.

51
Reconstruction As the number of species goes up, and the number of characters goes up, then conflicting data begins to appear. Then common sense and logic are insufficient for the job. The mutation rate may not be high enough to distinguish closely related species, – those near the bottom of the tree, but too high to make confident conclusions for reconstructing the top of the tree in order to connect distantly related species.

52
Reconstruction Also, deciding the relative lengths of the lines in the tree, – or the equivalent problem of deciding how high to put join the various lines, requires computations and a basis for making computations.

53
Reconstruction A simplification of the problem. There's a lot of information in the gene sequences, and it's difficult to analyze it all. One way to simplify things is to look at just pairs of species at a time. This will ignore some useful information, but enough will remain to do a pretty good job on reconstructing a phylogenetic tree, and the computations become simpler.

54
Reconstruction A simplification of the problem. When we look at two species, we have two sequences of characters, and the relevant measure is the number of differences in these two sequences, – a measure that we can interpret as the distance between the species. Algorithms that depend only on distances between species are called distance matrix algorims.

55
Reconstruction Distances between species. If two species have a small distance between them – (as measured by the number of differences in their character sequences), then they have a recent common ancestor; but if they are far apart, then their common ancestor is in the remote past.
, then they have a recent common ancestor; but if they are far apart, then their common ancestor is in the remote past..")
56
Reconstruction Distances between species. We can use the distance between the species as a measure of the distance in time since the species diverged. These two distances, 1. the number of character differences and 2. the time since divergence, will be approximately proportional when they're relatively small.

57
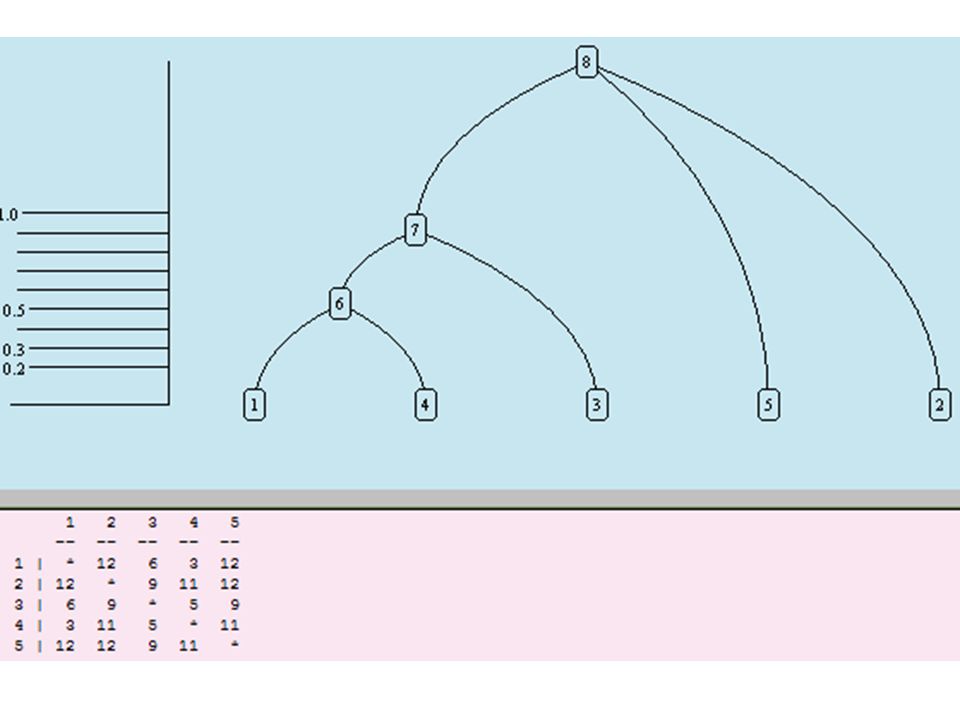
Reconstruction The difference matrix. Here is a model phylogenetic tree with six extant species alongside a matrix. This 6 by 6 matrix results from mutations of 40 irrelevant characteristics each with 4 alternate values. The mutation rate is uniform with a value of 100 mutations per 1000 time units, that is, 0.1 mutations per time unit.

58
Reconstruction The difference matrix. The (i,j)th entry in the matrix indicates how many of the 40 characters differ between species i and species j. If two species are not very distant in the tree, then there hasn't been much time for mutations to occur, so the entry in this matrix should be small.
th entry in the matrix indicates how many of the 40 characters differ between species i and species j. If two species are not very distant in the tree, then there hasn t been much time for mutations to occur, so the entry in this matrix should be small..")
59
Reconstruction The difference matrix. If two species are very distant, the entry in the matrix should be large, that is, close to 30, which is 3/4 of the number of characters. You won't see such large entries in the matrix unless you increase the mutation rate or the number of species.

60
Reconstruction The difference matrix. Note that the matrix is symmetric, that is, the (i,j)th entry is the same as the (j,i)th entry. Also, the entries along the diagonal are all 0, denoted here as *, since each (i,i)th entry indicates how many differences between the ith character sequence and itself, which, of course, is 0.
th entry is the same as the (j,i)th entry. Also, the entries along the diagonal are all 0, denoted here as *, since each (i,i)th entry indicates how many differences between the ith character sequence and itself, which, of course, is 0..")
61
Reconstruction Algorithms The problem Suppose all we know is how far apart the species are as measured by the number of differences in their characters, that is, the entries in the difference matrix. How can we reconstruct the phylogenetic tree? First, we can convert the differences to times.

62
Reconstruction Algorithms The problem The conversion is given by the formula

63
Reconstruction Algorithms Of course, we might not be able to reconstruct the actual tree, – since the mutations are random and need not reflect the actual distances between species. So a better question is: How can we reconstruct the most likely phylogenetic tree? We can come up with some promising algorithms that should give trees that aren't too far away from the most likely tree.

64
Reconstruction Algorithms A solution: the "minimum" reconstruction method. It seems reasonable that the two species that share the greatest number of characters are the most closely related. That is, the smallest entry in the mutation matrix indicates which two species diverged most recently. Also, the next smallest entry should indicate which two species diverged just before that. And so forth.

65
Reconstruction Algorithms A solution: the "minimum" reconstruction method. That's the idea of the algorithm, but it needs a little clarification. Suppose species 1 and 2 are closest with 4 differences in their characters, and species 1 and 3 are next closest with 6 differences. Then since we conclude that species 1 and 2 diverged most recently, it won't be species 1 and 3 that diverged just before that, rather it will be the ancestral species of 1 and 2 that diverged from species 3 just before that.

67
Reconstruction Algorithms Two other solutions: the "average" and "maximum" methods. They start out exactly the same by joining the two species that share the most characters. To explain these methods, suppose that species 1 and 2 are closest. Name their ancestral species as species 6. With the minimum method, we effectively determined that the distance between species 6 and any other species such as species 3 was the minimum of the distance from 1 to 3 and the distance from 2 to 3. With the maximum method, instead take the distance from species 6 to species 3 to be the maximum of these two distances. And, of course, for the average method, take the average of those two distances.

Similar presentations

>")

>")




 produce.>")











