Download presentation
Presentation is loading. Please wait.

1
FLEX* - REVIEW

2
Agenda Introduction Main concepts FlexE FlexS
Evaluation and Discussion

3
Introduction Methods for the prediction of binding properties of molecules to proteins. Classification by the amount of information available about the target protein

4
The general schema Modeling Algorithm
Ligand conformational flexibility Modeling Receptor-ligand interactions Scoring function Base selection Algorithm Base placement Incremental construction

5
The Ligand conformational flexibility
Approximated by a discrete set of conformations. rotatable single bond - modeled by a discrete set of preferred torsion angles from the MIMUMBA DB. Ring system - A set of ring conformations is computed with the program CORINA.

6
The model of receptor-ligand interactions
Modeled by a few special types of interactions hydrogen bonds metal acceptors bonds hydrophobic contacts

7
The model of protein-ligand interactions – Cont.
To each interaction group, we assign: Interaction types Interaction geometry ( center + surface)
")
8
Two groups interact if :
The centers of the groups lie approximately on the surface of the counter group. The interaction types are compatible The intermolecular interactions can be classified by the strength of their geometric constrains

9
Scoring function Estimates the free binding energy in the complex
The function is additive in the ligand atoms. match score contact score

10
Overall docking algorithm
Ligand fragmentation Select & Place a set of base fragments Construct the ligand by linking the remaining fragments.

12
Ligand fragmentation The ligand is decomposed into components by cutting at each acyclic bond. Fragmentation is a partition of the components of the molecule, such that every part, called fragment, is connected in the component tree.

13
Ligand fragmentation Good results are produced if the added fragments are small Every fragment, except for the base fragment consist of only one component.

14
Selecting a base fragment
The problem: Find a fragment which leads to low energy docking solution. Good base fragment properties: Placeability Specificity

15
Selecting a base fragment –Cont.
We look for fragments maximizing the function:

16
Rules for selecting a set of fragments
No base fragment is fully contained in another base fragment Each component occurs in at most two base fragments Each component in a base fragment must be either necessary for the connectivity of the fragment or it must have interaction centers.

17
The base placement algorithm
Goal: find positions of the base fragment in the active site such that sufficient number of favorable interactions between the fragment and the protein can occur simultaneously. Solution: pose clustering.

18
The base placement algorithm – Cont.
Preparation: Store all triangles of interaction points (IP) of the protein in a hash table. Find all the compatible fragment IP’s triangles. Clustering of the legal transformations
 of the protein in a hash table. Find all the compatible fragment IP’s triangles. Clustering of the legal transformations.")
19
The incremental construction algorithm
Input: solution set - set of partial placements with the ligands with the ligands constructed up to and including fragment i-1 Output: set of partial placements with the ligands with the ligands constructed up to and including fragment i

21
The complex construction algorithm – cont.
Adding the next fragment in all the possible conformations Reject extended placements that have strong overlap with the receptor or internal overlap with the ligand. Searching for new interactions Optimizing the positions of the partial ligand Selecting a new solution set Clustering the solution set

22
Optimizing the positions of the partial ligand
The placement is optimized when: New interactions are found. The placement contains slightly overlapping atoms between the receptor and the ligand.

23
Selecting a new solution set
Select k best-scoring solution Problem: the scoring values cannot be compared directly when different fragments are involved. Solution: estimate the score of the whole ligand, given a partial placement.

24
Clustering partial solutions
If no placement contains the other, the distance is infinity Otherwise, the distance is defined to be the RMSD of the intersecting atoms. A cluster is reduced to a single placement.

26
Protein flexibility - motivation
Induced fit – side chain or even backbone adjustments upon docking of different ligands to the same protein. Even small conformational changes are critical for docking applications e.g. if a rotate able bond prevents a ligand from binding in the correct position.

27
Protein flexibelity Main idea: describe the protein structure variations with a set of protein structures representing the flexibility, mutation or alternative models of a protein. The variability considered by flexE is defined by the differences within the given input structures.

28
United protein description
Data structure that administers the protein structures variations. Contains an ensemble of up to 30 possible conformation of the protein. Most of them are low energy conformations of the same protein.

29
United protein description - construction
Superposition Clustering Add picture - 8

30
Notation Component : all the atoms which belong to the same amino acid or mutation of the amino acid. Contains a backbone part and a side chain part Part : set of instances Instance : one of the alternative conformations.

31
United protein description - clustering
The superimposed structures are combined by clustering each part separately Complete linkage hierarchical cluster The clustered instances can be recombined to form new valid protein structures. (2) Regardless of the structures from which they originally stem. ===>>>>> the structures we dock into are not limited to the original ensemble structures.
 Regardless of the structures from which they originally stem. ===>>>>> the structures we dock into are not limited to the original ensemble structures.")
32
Incompatibility Two instances of the united protein description are incompatible if they cannot be realized simultaneously. Logical: two instances are alternative to each other Geometric: two logically compatible instances overlap Structural: two instances of the same chain are unconnected

33
Incompatibility graph
The last type of incoppatibility is aminly meant to avoid the combination of instances belonging to different loop comformations. [!!!!!!! why???????????????]

34
Incompatibility graph
The incompatibility is internally represented as a graph by using the instances as nodes and the connecting pairs of incompatible node by an edge. Valid protein structures correspond to independent set in the graph.

35
Selection of instances
The ligand is placed fragment by fragment into the active site by the incremental construction algorithm. After each construction step, all possible interactions are determined. Apply the scoring function for each instance. We chose the IS with the highest score.

36
chose the IS with the highest score.
Select the optimal IS The IS can be assembled from IS of the connected components. Apply a modified version of the Bron-Kerbosch algorithm.

37
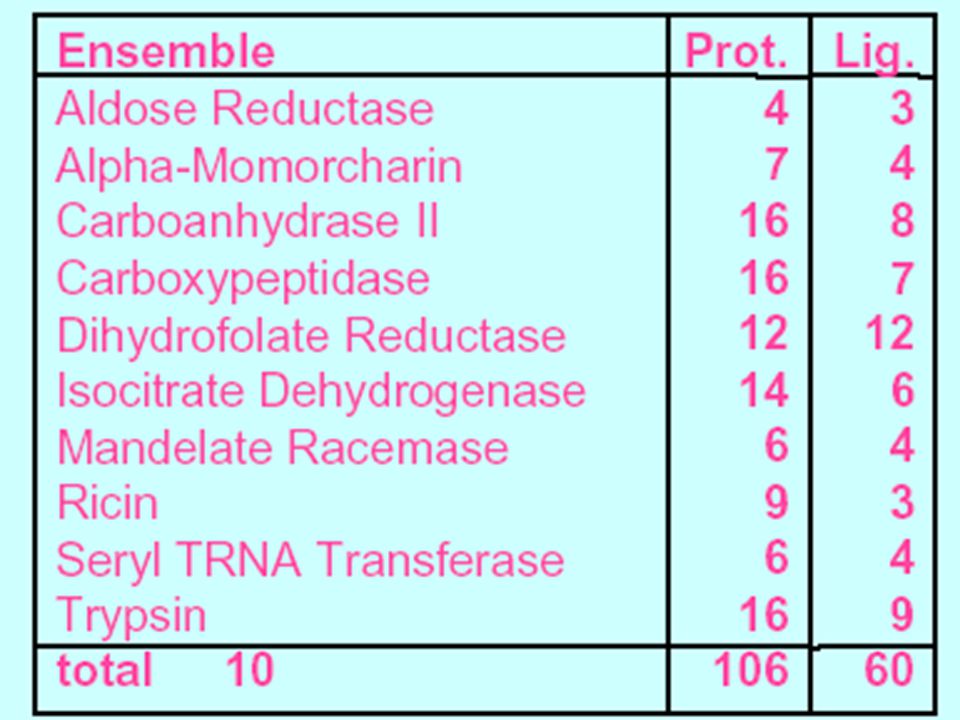
Evaluation FlexE was evaluated with ten protein structures ensembles containing 105 crystal structure from the PDB. The structures within the ensemble highly similar backbone trace Different conformations for several side chains.

39
Evaluation – Cont. FlexE finds a ligand position with RMSD below 2 A in 67% of the cases. Average CPU time for the incremental construction algorithm is 5.5 minutes.

41
Discussion The ensemble approach is able to cope with several side-chains conformations and even movements of loops. Motions of larger backbone segments or even domains movements are not covered by this approach.

43
flexS - motivation In drug design, often enough, no structural information about a particular receptor is available. Considerable number of different ligands are known together with their binding affinities towards the receptor.

44
flexS - overview A method for structurally superpositing pairs of ligands, approximating their putative binding site geometry. Main Applications ligand superpositioning Virtual Screening

45
Implementation in flexS
RigFit – fast rigid-body placement using Fourier space methods. Incremental construction Systematic parameter study

46
Two Base Placement Methods
Target: Place a rigid molecule fragment onto the reference ligand Combinatorial placement procedure Numerical placement procedure

47
RigFit Optimizes the common volume of two molecule expressed by various Gaussian functions associated to different physicochemical properties. Solves the combinatorial placement problem.

48
Variable Sequence Construction
The sequence in which fragments are added is selected dynamically depending on the actual placement. Effective in cases where the flexible test ligand partially extends beyond the reference ligand.

49
Dynamically selection of the next fragment
Each partial placement is associated with a list of candidate fragments. Evaluation of the next fragment considers: The amount of expected overlap with the reference The number of potential interaction in the candidate fragment The size of the substructure tree rooted at the candidate fragment.

50
Dynamically selection of the next fragment – Cont.
Nbus –number of buildup states. Deviation from the original sequence only if a better sequence is found If flexS exceeds Nbus upper limit, it returns to the original sequence

51
Evaluation The performance of the algorithm depends on the size of the superimposed ligands. In reproduction of 284 alignments, 60% reproduces with RMSD below A.

52
Questions?

53
Thank you!

54
Scoring & Selection strategy
Total score fo the partial ligand

55
FlexS Flow Test ligand Reference ligand fragmentation
Placement of the anchor molecule Add a fragment that adopts a discrete set of conformations

57
The physicochemical model
The conformational space of the ligand The model of protein-ligand interactions Scoring function

58
United protein description - superposition
Assumption: highly similar backbone traces -> superposition by fitting the backbone atoms of the particular structures. This procedure emphasizes the differences and improves the fitting in conserved regions of structures. [why ???] Superposition in two steps: 1. 2. Iteration of the first step. Pairs of atoms with a distance greater that a user defined threshold are ignored for the next step.

59
Surface and interaction geometries

Similar presentations









>")



 Observation:>")








![. Protein Structure Prediction [Based on Structural Bioinformatics, section VII]](/16/5116302/big_thumb.jpg ". Protein Structure Prediction [Based on Structural Bioinformatics, section VII]>")

 Prof. Chen Yu Zong Tel: 6874-6877>")
