Download presentation
Presentation is loading. Please wait.

1
Exploratory Failure Time Analysis and Copy Number Variation Inference Cheng Department of Biostatistics St. Jude Children’s Research Hospital

2
Outline Part IBackground Part IIExploratory Failure Time Analysis Part IIICopy Number Variation Inference

3
I. Background Nucleus, nucleotides, DNA, chromosomes, SNP SNP arrays Genome Wide Association Study (GWAS) Multiple tests Cause-specific failure and Competing risk Cumulative incidence function, Gray's test, Fine-Gray hazard rate regression model Censor at time competing event: OK for testing stochastic independence, biased for estimation
 Multiple tests Cause-specific failure and Competing risk Cumulative incidence function, Gray s test, Fine-Gray hazard rate regression model Censor at time competing event: OK for testing stochastic independence, biased for estimation.")
4
Animal CellAnimal Cell Organelles Nucleus Nucleolus Endoplasmic Reticulum Centriole Centrosome Golgi Cytoskeleton Cytosol Mitochondrion Secretory Vesicle Lysosome Peroxisome Vacuole

5
Nucleus Functions The cell nucleus is an organelle that forms the package for our genes and their controlling factors. Store genes on chromosomes Organize genes into chromosomes to allow cell division. Transport regulatory factors & gene products via nuclear pores Produce messages (messenger Ribonucleic acid or mRNA) that code for proteins Produce ribosomes in the nucleolus Organize the uncoiling of DNA to replicate key genes
 that code for proteins Produce ribosomes in the nucleolus Organize the uncoiling of DNA to replicate key genes.")
6
Chromosome inside nucleus What is a chromosome? – In the nucleus of each cell, the DNA molecule is packaged into thread-like structures called chromosomes. – Each chromosome is made up of DNA tightly coiled many times around proteins called histones that support its structure. DNA = deoxyribonucleic acid

7
Human chromosomes In humans, each cell normally contains 23 pairs of chromosomes, for a total of 46. Twenty-two of these pairs, called autosomes, look the same in both males and females. The 23rd pair, the sex chromosomes, differ between males and females. – Females have two copies of the X chromosomeX chromosome – males have one X and one Y chromosome.Y chromosome

8
Chromosome Structure Each chromosome has a constriction point called the centromere, which divides the chromosome into two sections, or “arms.” The short arm of the chromosome is labeled the “p arm.” The long arm of the chromosome is labeled the “q arm.” Each chromosome has two chromatids as a result of duplication of the DNA which took place during interphase. The two chromatids are linked together at a centromere.

9
DNA structure DNA is a double-stranded molecule twisted into a helix (think of a spiral staircase). Each spiraling strand, comprised of a sugar-phosphate backbone and attached bases, is connected to a complementary strand by non-covalent hydrogen bonding between paired bases. The bases are adenine (A), thymine (T), cytosine (C) and guanine (G).
, thymine (T), cytosine (C) and guanine (G)..")
10
SNPs occur in human DNA at a frequency of one every 1,000 bases. These variations can be used to track inheritance in families. Genetic code is specified by the four nucleotide "letters" A (adenine), C (cytosine), T (thymine), and G (guanine). A Single Nucleotide Polymorphism (SNP) is a change of a single nucleotide, such as an T, replaces one of the other three nucleotide letters -- A, C, or G, within a person's DNA sequence.
, C (cytosine), T (thymine), and G (guanine). A Single Nucleotide Polymorphism (SNP) is a change of a single nucleotide, such as an T, replaces one of the other three nucleotide letters -- A, C, or G, within a person s DNA sequence..")
11
Genomic Sequence 5´3´ SNP T / G SNP probe = 25 bases Allele ‘A’ Perfect Match Mismatch Perfect Match Mismatch Allele ‘B’ SNP Array Design Quartet

12
Hundreds of Millions of Pixel Intensities…..

13
Genotype Calling AAABBB

14
Genome Wide Association Study (GWAS) Typically 400,000 to 900,000 SNPs are investigated in a single study Number of subjects in a study typically ranges from a few hundreds to 20,000 Each SNP takes three possible (generic) values “AA”, “AB”, “BB”, often coded as 0, 1, 2 Each SNP in each individual has a unique value, which is one of 0, 1, or 2 A small number of phenotypes: disease status (yes/no), or quantitative trait This lecture: time to a cause-specific failure n subjects, n observed trait values Y 1, …, Y n, n observed SNP values for the ith SNP X i1, …, X in Inference (Test) for stochastic dependence of the ith SNP with the trait based on the dataset (X ij, Y j ), j=1,…,n; do this for each SNP; thus many tests of the null hypothesis of stochastic independence.
 Typically 400,000 to 900,000 SNPs are investigated in a single study Number of subjects in a study typically ranges from a few hundreds to 20,000 Each SNP takes three possible (generic) values AA , AB , BB , often coded as 0, 1, 2 Each SNP in each individual has a unique value, which is one of 0, 1, or 2 A small number of phenotypes: disease status (yes/no), or quantitative trait This lecture: time to a cause-specific failure n subjects, n observed trait values Y 1, …, Y n, n observed SNP values for the ith SNP X i1, …, X in Inference (Test) for stochastic dependence of the ith SNP with the trait based on the dataset (X ij, Y j ), j=1,…,n; do this for each SNP; thus many tests of the null hypothesis of stochastic independence.")
15
Massive Multiple Tests “Genome-wide significance” Bonferroni-type adjustment: Declare statistical significance if P≤10 -7 (0.05/500K) FDR and q value Benjamini, Y. and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. JRSS-B, 57, 289–300. Storey, J. D., Taylor, J. and Siegmund, D. (2003). Strong control, conservative point estimation and simultaneous conservative consistency of false discovery rates: a unified approach JRSS-B, 66, 187–205. Profile information criteria Cheng, C., Pounds, S., Boyett, J. M. et al (2004). Statistical significance threshold criteria for analysis of microarray gene expression data. Statistical Applications in Genetics and Molecular Biology 3, Article 36. URL //www.bepress.com/sagmb/vol3/iss1/art36 Cheng, C (2006) An adaptive significance threshold criterion for massive multiple hypotheses testing. IMS Lecture Notes - Monograph Series 2nd Lehmann Symposium – Optimality 49, 51–76
. Controlling the false discovery rate: a practical and powerful approach to multiple testing. JRSS-B, 57, 289–300. Storey, J. D., Taylor, J. and Siegmund, D. (2003). Strong control, conservative point estimation and simultaneous conservative consistency of false discovery rates: a unified approach JRSS-B, 66, 187–205. Profile information criteria Cheng, C., Pounds, S., Boyett, J. M. et al (2004). Statistical significance threshold criteria for analysis of microarray gene expression data. Statistical Applications in Genetics and Molecular Biology 3, Article 36. URL // Cheng, C (2006) An adaptive significance threshold criterion for massive multiple hypotheses testing. IMS Lecture Notes - Monograph Series 2nd Lehmann Symposium – Optimality 49, 51–76.")
16
Cause-specific failure and competing risk Alive Relapse 2 nd Cancer Die in remission Failure type 1 (of interest) Failure type 2 (competing risk/event) Failure type 3 (competing risk) Klein, J. P. (2010) Competing risks. WIREs Comp Stat, www.wiley.com/wires/compstats, DOI: 10.1002/wics.83
 Competing risks. WIREs Comp Stat, DOI: /wics.83.")
17
Cumulative incidence function (CIN) (T, δ); F j (t)=Pr(T ≤ t and δ=j) Gray’s test: Compare CIN across K groups Analog of weighted log-rank test Gray, R. J. (1988) A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann. Statist. 16, 1141-1154. Fine-Gray’s CIN hazard rate regression model Analog of Cox’s hazard rate regression model Fine, J. P., Gary, R.J. (1999) A proportional hazards model for the subdistribution of a competing risk. JASA, 94, 496-509. Censor at the time of competing event
 A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann. Statist. 16, Fine-Gray’s CIN hazard rate regression model Analog of Cox’s hazard rate regression model Fine, J. P., Gary, R.J. (1999) A proportional hazards model for the subdistribution of a competing risk. JASA, 94, Censor at the time of competing event.")
18
II. Exploratory Failure Time Analysis Large-scale Genomic Association Analysis o Feature (variable) screening and feature extraction A Motivating Example from a GWAS Correlation Profile Test (CPT) o Hypotheses o Correlation profile function o CPT statistic o Hybrid permutation test of significance A Simulation Study: Strength and Weakness Example: Analysis of SNPs on Chromosome 9 Summary and Remarks Feature Extraction (sparse regression) Example: “Prognostic” Gene (RNA) expression Summary and remarks
 screening and feature extraction A Motivating Example from a GWAS Correlation Profile Test (CPT) o Hypotheses o Correlation profile function o CPT statistic o Hybrid permutation test of significance A Simulation Study: Strength and Weakness Example: Analysis of SNPs on Chromosome 9 Summary and Remarks Feature Extraction (sparse regression) Example: Prognostic Gene (RNA) expression Summary and remarks.")
19
Large-scale Genomic Association Analysis Feature (variable) screening – Find individual genomic features (factor/predictor variables) associated with one or more phenotypes (response variables) GWAS – Association: stochastic dependence – Parametric/semi-parametric approaches: linear models, GLMs, hazard rate (Cox) regression Feature extraction – Find (linear) combinations (or sets) of genomic features (variables) associated with one or more phenotypes – Determine sets of variables using biological knowledge (gene signaling pathways, functional/ontology groups, etc.): GSEA – Variable/Model selection methods: ridge regression, LASSO, SCAD, SEAMLESS, sparse regression
 screening – Find individual genomic features (factor/predictor variables) associated with one or more phenotypes (response variables) GWAS – Association: stochastic dependence – Parametric/semi-parametric approaches: linear models, GLMs, hazard rate (Cox) regression Feature extraction – Find (linear) combinations (or sets) of genomic features (variables) associated with one or more phenotypes – Determine sets of variables using biological knowledge (gene signaling pathways, functional/ontology groups, etc.): GSEA – Variable/Model selection methods: ridge regression, LASSO, SCAD, SEAMLESS, sparse regression")
20
A Motivating Example GWAS to screen SNP markers for risk of relapse in childhood leukemia patients AAABBB X012 Relapse7000 Comp. Event2400 Censored585119

21
A Motivating Example Need: a more omnibus and algorithmically robust test procedure AAABBB X012 Relapse7000 Comp. Event2400 Censored585119 PCoeff. (s.e.) Cox Regression Test of coeff.1-16.4 (2901) LR test0.0458 R gives a warning Fine-Gray Regression JASA 1999 94(446):496-509 Test of coeff.0-11.4 (0.334) LR test0.0488 Gray's Test0.3542 Ann. Statist. 1988 16(3):1141-1154 Jung's Test0.6607 Statist. Medicine 2005 24:3077-3088
 Cox Regression Test of coeff (2901) LR test R gives a warning Fine-Gray Regression JASA (446): Test of coeff (0.334) LR test Gray s Test Ann. Statist (3): Jung s Test Statist. Medicine :")
22
Model, Null and alternative hypotheses (classical survival setting) Correlation Profile Test (CPT)
 Correlation Profile Test (CPT)")
23
Sample correlation profile function observed event point process of individual i Can do rank transformation for continuous X

24
Correlation Profile Test (CPT)
")
25
CPT statistic, hybrid permutation test

26
Back to the SNP Example AAABBB X012 Relapse7000 Comp. Event2400 Censored585119 PCoeff. (s.e.) Cox Regression Test of coeff.1-16.4 (2901) LR test0.0458 R gives a warning Fine-Gray Regression JASA 1999 94(446):496-509 Test of coeff.0-11.4 (0.334) LR test0.0488 Gray's Test0.3542 Ann. Statist. 1988 16(3):1141-1154 Jung's Test0.6607 Statist. Medicine 2005 24:3077-3088 CPT0.2582 Test stat is negative
 Cox Regression Test of coeff (2901) LR test R gives a warning Fine-Gray Regression JASA (446): Test of coeff (0.334) LR test Gray s Test Ann. Statist (3): Jung s Test Statist. Medicine : CPT Test stat is negative.")
27
A Simulation Study A model mimicking the SNP example Generate X: Pr(X=0)=0.98, Pr(X=1)=0.015, Pr(X=2)=0.005 Generate Censor Time T C ~ Exp(0.2) Generate failure indicator I F |X ~ Bernoulli(π F ); π F = 0.2exp{-θ(X-2)} If I F = 1, generate Failure Time T F |X ~ LogNormal(βX,1) else set T F = ∞ Generate competing risk indicator I R ~ Bernoulli(0.1) If I R = 1, generate Competing Failure Time T R ~ Unif(0,7) else set T R = ∞ Observed Failure Time T = min{T C T F T R } Repeat the above n times to simulate n individuals
=0.98, Pr(X=1)=0.015, Pr(X=2)=0.005 Generate Censor Time T C ~ Exp(0.2) Generate failure indicator I F |X ~ Bernoulli(π F ); π F = 0.2exp{-θ(X-2)} If I F = 1, generate Failure Time T F |X ~ LogNormal(βX,1) else set T F = ∞ Generate competing risk indicator I R ~ Bernoulli(0.1) If I R = 1, generate Competing Failure Time T R ~ Unif(0,7) else set T R = ∞ Observed Failure Time T = min{T C T F T R } Repeat the above n times to simulate n individuals")
28
A Simulation Study A model mimicking the SNP example 0.05 0.01 CPTFGJungGray CPTFGJungGray Null θ=0, β=0 0.0486 0.00215 0.1953 0.0040 0000 0.0711 0.0007 0.0199 0.0014 0.1658 0.0037 0000 0.0281 0.0003 θ=0 β=0.5 0.09 0.0028 0.17 0.0038 0000 0.096 0.0029 0.007 0.0008 0.069 0.0025 0000 0.016 0.0012 θ=0.5 β=1.2 0.453 0.0050 0.502 0.0050 0000 0.277 0.0045 0.04 0.0020 0.189 0.0039 0000 0.05 0.0022 θ=0.8 β=0 0.67 0.0047 0.802 0.0040 0.004 0.0006 0.807 0.0039 0.378 0.0048 0.495 0.0050 0000 0.565 0.0050 θ=0.8 β=1.2 0.981 0.0014 0.99 0.0010 0.034 0.0018 0.967 0.0018 0.875 0.0033 0.867 0.0034 0.003 0.0005 0.869 0.0034 Pwr est. s.e.

29
A Simulation Study 0.01 0.005 CPTCoxJung CPTCoxJung Null0.0088 0.0009 0.0112 0.0011 0.0096 0.0010 0.0047 0.0007 0.0067 0.0008 0.0053 0.0007 β=0.50.093 0.0029 0.173 0.0038 0.117 0.0032 0.066 0.0025 0.123 0.0033 0.082 0.0027 β=0.80.359 0.0048 0.617 0.0049 0.518 0.0050 0.268 0.0044 0.524 0.0050 0.436 0.0050 Exact Proportional Hazard, continuous predicator

30
A Simulation Study 0.01 0.005 CPTCoxJung CPTCoxJung β=0.5 n=300 0.149 n=400 0.181 n=200 0.173 n=200 0.117 n=300 0.094 n=400 0.127 n=200 0.123 n=200 0.082 β=0.8 n=300 0.537 n=400 0.730 n=200 0.617 n=200 0.518 n=300 0.449 n=400 0.655 n=200 0.524 n=200 0.436 Exact Proportional Hazard, continuous predicator

31
A Simulation Study 0.01 0.005 CPTFGJungCPTFGJung β 1 =0 β=0 0.0082 0.0009 0.0159 0.0012 0.0124 0.0011 0.0039 0.0006 0.0096 0.0010 0.0052 0.0007 β 1 =1 β=0 0.193 0.0039 0.019 0.0014 0.027 0.0016 0.13 0.0033 0.011 0.0010 0.016 0.0012 β 1 =2 β=0 0.361 0.0048 0.031 0.0017 0.045 0.0021 0.254 0.0044 0.022 0.0015 0.025 0.0016 β 1 =3 β=0 0.399 0.0049 0.054 0.0022 0.076 0.0026 0.302 0.0046 0.033 0.0018 0.046 0.0021 β=0.6 β 1 =0 0.325 0.0047 0.236 0.0042 0.201 0.0040 0.231 0.0042 0.165 0.0037 0.125 0.0033 β=1.2 β 1 =0 0.631 0.0048 0.698 0.0046 0.596 0.0049 0.502 0.0050 0.587 0.0049 0.488 0.0050 Continuous predictor, deviation from proportional hazard

32
0.01 0.005 CPTFGJungGray CPTFGJungGray Null θ=0, β=0 0.0088 0.0009 0.0109 0.0010 0.005 0.0007 0.0044 0.0006 0.0063 0.0008 0.0019 0.0004 θ=0 β=0.6 0.172 0.0038 0.057 0.0023 0.025 0.0016 0.061 0.0024 0.121 0.0033 0.043 0.0020 0.016 0.0012 0.041 0.0020 θ=0 β=1.2 0.551 0.0050 0.36 0.0048 0.211 0.0041 0.273 0.0044 0.471 0.0050 0.286 0.0045 0.142 0.0035 0.214 0.0041 θ=0.25 β=0 0.059 0.0024 0.099 0.0030 0.057 0.0023 0.075 0.0026 0.037 0.0020 0.076 0.0027 0.039 0.0019 0.061 0.0024 θ=0.25 β=1.2 0.862 0.0034 0.708 0.0045 0.569 0.0050 0.621 0.0048 0.801 0.0040 0.632 0.0048 0.464 0.0050 0.538 0.0050 θ=0.5 β=0 0.314 0.0046 0.525 0.0050 0.375 0.0048 0.441 0.0050 0.236 0.0042 0.452 0.0050 0.288 0.0045 0.355 0.0048 θ=0.5 β=0.6 0.895 0.0031 0.831 0.0037 0.713 0.0045 0.782 0.0041 0.847 0.0036 0.76 0.0043 0.618 0.0049 0.712 0.0045 A Simulation Study Ordinal predictor, deviation from proportional hazard Opposite scenario of the SNP example AA AB BB

33
Example: Germline SNPs on Chr 9 and risk of relapse in childhood Acute Lymphoblastic Leukemia (ALL) 21,909 SNPs on Chr 9 obtained by Affy 100K and 500K SNP arrays were tested for association with relapse of childhood ALL Alive Relapse 2 nd Cancer Die in remission Failure type 1 (of interest) Failure type 2 (competing risk/event) Failure type 3 (competing risk)
 21,909 SNPs on Chr 9 obtained by Affy 100K and 500K SNP arrays were tested for association with relapse of childhood ALL Alive Relapse 2 nd Cancer Die in remission Failure type 1 (of interest) Failure type 2 (competing risk/event) Failure type 3 (competing risk)")
34
Example: Germline SNPs on Chr 9 and risk of relapse in childhood Acute Lymphoblastic Leukemia (ALL) n=707 subjects from two most recent clinical trial at SJCRH 21,909 SNPs CPT test performed on each SNP, with 200 permutations in the hybrid permutation test Significance determined by the profile info criteria Ip (Cheng et al. 2000); 200 SNPs were considered statistically significant, estimated FDR=48.7%
; 200 SNPs were considered statistically significant, estimated FDR=48.7%.")
37
SNPPval.CPTAnnotation SNP_A-42168036.44E-06C9orf82.downstream.461318.AFFY SNP_A-21422237.87E-05TMC1.upstream.48246.AFFY//ZFAND5.upstream.108579.AFFY SNP_A-18787198.80E-05PTPRD.In_gene.5000.5kRuleLD SNP_A-42012960.000126717DBC1.In_gene.5000.5kRuleLD SNP_A-42549750.000131144JMJD2C.downstream.206626.AFFY SNP_A-22896680.000138093GLIS3.In_gene.5000.5kRuleLD SNP_A-18479560.000140329RFX3.downstream.268660.AFFY SNP_A-19959350.000169608C9orf150.downstream.49950.AFFY SNP_A-19962760.000182668ELAVL2.In_gene.5000.5kRuleLD SNP_A-22020300.000216207C9orf93.downstream.130169.AFFY SNP_A-21009560.000254439C9orf82.downstream.513396.AFFY SNP_A-20985140.000337669BNC2.In_gene.5000.5kRuleLD SNP_A-22284600.000398633GLIS3.upstream.6282.AFFY SNP_A-42525170.00040459TUSC1.downstream.286633.AFFY SNP_A-19175900.000430395PTPRD.In_gene.5000.5kRuleLD SNP_A-20527520.000432044DMRT1.In_gene.5000.5kRuleLD SNP_A-20610980.000448184C9orf94.In_gene.5000.5kRuleLD SNP_A-17865170.000478508GSN.In_gene.5000.5kRuleLD SNP_A-23049200.000575635UBE2R2.In_gene.5000.5kRuleLD SNP_A-19023720.00057778GSN.In_gene.5000.5kRuleLD SNP_A-22013000.000588075ABL1.In_gene.5000.5kRuleLD SNP_A-22382680.000592182PCSK5.upstream.10183.AFFY SNP_A-18301830.000613767TUSC1.downstream.181857.AFFY

38
ρ^(t j ), j=1, …, J=9 Test stat = -3.478
, j=1, …, J=9 Test stat =")
39
AA 5.1% AB28.7% BB66.2% P Gary’s test0.0451 Fine-Gray regression0.0380; coeff=-0.3905

40
ABL1 Gene Germline SNP AAABBBTot 36 (0.051)201 (0.287)464 (0.662)701 (1.00) A273 (0.195) B1129 (0.805) AAABBB T13B intermediate/high risk122775 (0.152) T13B Low risk73367 (0.065) T15 standard/high risk1174161 (0.047) T15 Low risk667161 (0.026)
201 (0.287)464 (0.662)701 (1.00) A273 (0.195) B1129 (0.805) AAABBB T13B intermediate/high risk (0.152) T13B Low risk73367 (0.065) T15 standard/high risk (0.047) T15 Low risk (0.026)")
41
Extension to Recurrent Events Model, Null and alternative hypotheses Multiple event times # events occurred ≤ t

42
Extension to Recurrent Events N = # events occurred ≤ t N

43
Summary and Remarks Correlation Profile Test: – Computationally more robust – More omnibus: covers certain deviations from the semi-parametric hazard regression model – Highly competitive with other non-parametric procedures (Gray’s test, Jung’s test) – Relative deficiency vs. Cox model under PH ?? – Extension to recurrent-event phenotypes – Informative censoring in the presence of competing risk

44
Feature Extraction (Sparse regression) Identify (linear) combinations of covariate variables that are associated with the failure phenotype
 Identify (linear) combinations of covariate variables that are associated with the failure phenotype")
45
Feature Extraction (Sparse regression) Sparse regression by the General Path seeking (GPS) algorithm (Friedman 2008) Exploratory failure time analysis by weighted least square -- the association criteria The modified GPS algorithm to find a solution A small simulation study Example: Gene (RNA) expression “prognostic” for relapse of childhood ALL
 Sparse regression by the General Path seeking (GPS) algorithm (Friedman 2008) Exploratory failure time analysis by weighted least square -- the association criteria The modified GPS algorithm to find a solution A small simulation study Example: Gene (RNA) expression prognostic for relapse of childhood ALL")
46
Sparse Regression by General Path Seeking (GPS, Friedman 2008) http://www-stat.stanford.edu/~jhf//ftp/GPSpub.pdf General Setup http://www-stat.stanford.edu/~jhf//ftp/GPSpub.pdf Lasso (Tibshirani 1996), grouped lasso (Yuan and Lin 2006), SCAD (Fan and Li 2001) Elastic net (Zuo and Hastie 2005) SEAL (Xihong Lin, 2009 JSM)
 General Setup Lasso (Tibshirani 1996), grouped lasso (Yuan and Lin 2006), SCAD (Fan and Li 2001) Elastic net (Zuo and Hastie 2005) SEAL (Xihong Lin, 2009 JSM)")
47
Feature Extraction (Sparse regression) The general GPS algorithm
 The general GPS algorithm")
48
Feature Extraction (Sparse regression) Exploratory failure time analysis: setup
 Exploratory failure time analysis: setup")
49
Feature Extraction (Sparse regression) Association criteria: Penalized weighted least square
 Association criteria: Penalized weighted least square")
50
Feature Extraction (Sparse regression) The power penalty function |β| γ, 0<γ≤1 γ =1 γ =0.5 γ =0.0001
 The power penalty function |β| γ, 0<γ≤1 γ =1 γ =0.5 γ =0.0001")
51
Feature Extraction (Sparse regression) The modified GPS algorithm
 The modified GPS algorithm")
52
Feature Extraction (Sparse regression) Gradient descent with a fixed step size; searches the solution along a sequence of increasing values of the penalty parameter λ, thus no need to use CV or GCV type criteria to determine λ. Initial value: all β’s are set to zero Each iteration modifies just one of the m dimensions, criteria to choose which dimension to update involves the gradient of the association criteria and penalty function Need to modify for this particular application – Relatively large step size Δν : 0.01 (sometimes) – Stopping rule: Stop if the size of the gradient vector does not change by more than Δν from the previous iteration or pre-specified max number of iterations is reached
 – Stopping rule: Stop if the size of the gradient vector does not change by more than Δν from the previous iteration or pre-specified max number of iterations is reached.")
53
Feature Extraction (Sparse regression) A small simulation study Simulation model: Proportional hazard
 A small simulation study Simulation model: Proportional hazard")
54
Characteristic of SolutionFreq.% X1 & X2 no false + (perfect)275.4 X1 & X2 w/ false +408 X2 only no false +22344.6 X2 only w/ false +19939.8 X1 only no false +20.4 X1 only w/ false +10.2 None (all false +, worst)81.6 TOTAL500100 Feature Extraction (Sparse regression) A small simulation study Performance assessment
275.4 X1 & X2 w/ false +408 X2 only no false X2 only w/ false X1 only no false X1 only w/ false None (all false +, worst)81.6 TOTAL Feature Extraction (Sparse regression) A small simulation study Performance assessment")
55
Feature Extraction (Sparse regression) A small simulation study Performance assessment #non-zeroFreq. (%)X1-onlyX2-onlyBothnone 1230 (46)222305 2132 (26.4)0103272 370 (14)154141 431 (6.2)021100 519 (3.8)01090 610 (2)0550 77 (1.4)0520 81 (0.2)0100
X1-onlyX2-onlyBothnone 1230 (46) (26.4) (14) (6.2) (3.8) (2) (1.4) (0.2)0100.")
56
Feature Extraction (Sparse regression) A small simulation study Performance assessment R=# of non-zeros in solution V=# of incorrect non-zeros in solution FDR = E(V/R|R>0) Estimated FDR = 0.2984 s.e. = 0.0142 99% CI (0.2618, 0.3350)
.")
57
Feature Extraction (Sparse regression) An Example: Gene (RNA) expression in ALL and risk of relapse Affymetrix U133A GeneChip n=287 Arrays (subjects) m=22,278 Probesets Two clinical variables: Age group at Dx, lineage Intercept term Total number of variables = m+3
 An Example: Gene (RNA) expression in ALL and risk of relapse Affymetrix U133A GeneChip n=287 Arrays (subjects) m=22,278 Probesets Two clinical variables: Age group at Dx, lineage Intercept term Total number of variables = m+3")
58
Feature Extraction (Sparse regression) Example (Cont.) Run parameters: – step size = 6 x 10 -4, γ = 0.01 Initial values: – Coeff. of Age = 6 x 10 -4 – Coeff of Lineage = 6 x 10 -4 – All others set to 10 -8

59
Top meaningful findings VariableCoeffGene Age.DX0.183 Lineage0.1302 212869_x_at0.003TPT1; (similar to) tumor protein; translationally controlled 201288_at0.0006RhoGDI2; plays a role in apoptosis; may be a marker for tumor progression in gastric and breast cancer; literature not consistent
 tumor protein; translationally controlled _at0.0006RhoGDI2; plays a role in apoptosis; may be a marker for tumor progression in gastric and breast cancer; literature not consistent")
60
Summary and Remarks The sparse regression approach to exploratory survival analysis – Step size of descent is crucial – Newton-type descent more adaptive, maybe better, how to incorporate? – Stopping criteria: change in gradient vector? size of the gradient vector? – Other association criteria: -log likelihood by Logistic, Probit, Poisson etc. links – “Oracle” property of the solution? “Accuracy” of the solution? in asymptopia

61
III. Copy Number Variation Inference Cell division DNA Copy Number Variation (CNV) Use SNP array signals to infer CNV Reference signal alignment: example Reference signal alignment procedure Recent development Examples
 Use SNP array signals to infer CNV Reference signal alignment: example Reference signal alignment procedure Recent development Examples.")
62
Cell division: How cell grow and divide Mitosis: The process in somatic cell division by which the nucleus divides Mitosis: Meiosis: The process of cell division in sexually reproducing organisms that reduces the number of chromosomes in reproductive cells from diploid to haploid, leading to the production of gametes in animals and spores in plants Meiosis:

63
Mitosis: Prophase Prophase is the first stage of cell division, the cell prepares itself for division. The nucleus swells, and chromosomes become visible. Each chromosome has two chromatids as a result of duplication of the DNA which took place during interphase. The two chromatids are linked together at a centromere. The centrosome (2 centrioles) duplicates into 2 diplosomes, and each diplosome, or aster moves toward opposite poles of the nucleus.
 duplicates into 2 diplosomes, and each diplosome, or aster moves toward opposite poles of the nucleus..")
64
Mitosis: Metaphase Microtubules assemble, and form a network (the spindle fibres). The chromosomes move towards the equator of the cell, where they are visible. This is the phase in which morphological studies of chromosomes are carried out, often for clinical purposes.

65
Mitosis: Anaphase The two sister chromatids separate. Each one migrates to opposite ends of the cell. So each daughter cell has an identical complement of chromosomes. The nuclear membrane has disappeared at this stage. The cell membrane expands as the cell itself elongates. The diameter of the cell decreases at the equator.

66
Mitosis: Telophase A new membrane forms around the new nuclei and two cells are quickly formed. The chromatid, now called a chromosome, uncoils, and the nucleolus becomes visible again. Each cell contains a pair of chromosomes (2n chromosomes)
.")
67
Meiosis The process of meiosis essentially involves two cycles of division, involving a gamete mother cell (diploid cell) dividing and then dividing again to form 4 haploid cells. These can be subdivided into four distinct phases which are a continuous process 1st Division – Prophase - Homologous chromosomes in the nucleus begin to pair up with one another and then split into chromatids (one half of a chromosome) where cross over can occur. Cross over can increase genetic variation. – Metaphase - Chromosomes line up at the equator of the cell, where the sequence of the chromosomes lined up is at random, through chance, increasing genetic variation via independent assortment. – Anaphase - The homologous chromosomes move to opposing poles from the equator – Telophase - A new nuclei forms near each pole alongside its new chromosome compliment. – At this stage two haploid cells have been created from the original diploid cell of the parent. 2nd Division – Prophase II - The nuclear membrane disappears and the second meiotic division is initiated. – Metaphase II - Pairs of chromatids line up at the equator – Anaphase II - Each of these chromatid pairs move away from the equator to the poles via spindle fibres – Telophase II - Four new haploid gametes are created that will fuse with the gametes of the opposite sex to create a zygote.
 where cross over can occur. Cross over can increase genetic variation. – Metaphase - Chromosomes line up at the equator of the cell, where the sequence of the chromosomes lined up is at random, through chance, increasing genetic variation via independent assortment. – Anaphase - The homologous chromosomes move to opposing poles from the equator – Telophase - A new nuclei forms near each pole alongside its new chromosome compliment. – At this stage two haploid cells have been created from the original diploid cell of the parent. 2nd Division – Prophase II - The nuclear membrane disappears and the second meiotic division is initiated. – Metaphase II - Pairs of chromatids line up at the equator – Anaphase II - Each of these chromatid pairs move away from the equator to the poles via spindle fibres – Telophase II - Four new haploid gametes are created that will fuse with the gametes of the opposite sex to create a zygote..")
68
Meiosis

69
Meiosis vs. Mitosis MITOSIS (In somatic cells)MEIOSIS (In reproductive cells ) One single division of the mother cell (m) results in two daughter cells (d) Two divisions of the mother cell result in four meiotic products (p) The number of chromosomes per nucleus remains the same after division The meiotic products contain a haploid (n) number of chromosomes, in contrast to the 2n mother cell Chromosomal re-distribution Chromosomal change (cross over) and re-distribution Gain/duplication or loss of DNA can occur in either process, resulting in deviations from the normal, 2-copy state of the chromomes or segments on chromosomes.
MEIOSIS (In reproductive cells ) One single division of the mother cell (m) results in two daughter cells (d) Two divisions of the mother cell result in four meiotic products (p) The number of chromosomes per nucleus remains the same after division The meiotic products contain a haploid (n) number of chromosomes, in contrast to the 2n mother cell Chromosomal re-distribution Chromosomal change (cross over) and re-distribution Gain/duplication or loss of DNA can occur in either process, resulting in deviations from the normal, 2-copy state of the chromomes or segments on chromosomes..")
70
Oncogenesis DNA gain: Excess of genes promoting cell division and proliferation DNA loss: Loss of gene functions regulating cell cycles, such as signaling apoptosis. DNA loss: Loss of functions necessary for proper lineage differentiation

71
Karyotyping and Complex CNV patterns in tumor genomes An assay technology to assess gains/losses of DNA; now routinely performed at diagnosis of childhood leukemia; not so readily available for solid tumors 67,XXY,-3,+8,-9,-16,-17,+20/66,idem,del(X)(p22.1),-8,del(10)(q22q26),-20,+mar 1-2(3)3(2)4-7(3)8(4)9(2)10-15(3)16-17(2)18-19(3)20(4)21-22(3)X(2) 6 2 12 4 2 18 4 6 4 6 2 total=66 +Y = 67
(p22.1),-8,del(10)(q22q26),-20,+mar 1-2(3)3(2)4-7(3)8(4)9(2)10-15(3)16-17(2)18-19(3)20(4)21-22(3)X(2) total=66 +Y = 67")
72
Contemporary technology: Array Comparative Genome Hybridization (aCGH)
")
73
Use SNP array signals to infer CNV Goal: Infer loss/gain -- qualitative

74
Use SNP array signals to infer CNV Importance of normalization Reason for single-array reference alignment: example from paper

75
Use SNP array signals to infer CNV Motivation/Reason for single-array reference alignment

76
Use SNP array signals to infer CNV Basic algorithm (Pounds et al. 2009) 1. Select a chromosome that is most likely in the 2-copy (diploid) state – make an educated guess 2. Use the empirical distribution (EDF) of the signals of the markers on this chromosome to transform all marker signals into the unit interval (0,1) via the probability-integral (quantile) transformation 3. Map the above transformed data into a known, convenient target distribution (e.g., N(0,1)); this produces the reference-aligned signals 4. Perform CNV segmentation using the above reference- aligned signals. Note after Step 3 the empirical distr. of reference markers is essentially the same as the target distribution (N(0,1))
 1. Select a chromosome that is most likely in the 2-copy (diploid) state – make an educated guess 2. Use the empirical distribution (EDF) of the signals of the markers on this chromosome to transform all marker signals into the unit interval (0,1) via the probability-integral (quantile) transformation 3. Map the above transformed data into a known, convenient target distribution (e.g., N(0,1)); this produces the reference-aligned signals 4. Perform CNV segmentation using the above reference- aligned signals. Note after Step 3 the empirical distr. of reference markers is essentially the same as the target distribution (N(0,1)).")
77
HOW TO SELECT THE REFERENCE CHROMOSOME? Pounds et al. (2009) Cytonormalization – Utilize karyotype data: select a chromosome not implied as abnormal by karyotyping Algorithmic selection – Select a chromosome that appears most likely be in the diploid state based on a set of statistics, such the percentage of heterozygous calls, joint behavior of signal mean and standard deviation (details in paper).
 Cytonormalization – Utilize karyotype data: select a chromosome not implied as abnormal by karyotyping Algorithmic selection – Select a chromosome that appears most likely be in the diploid state based on a set of statistics, such the percentage of heterozygous calls, joint behavior of signal mean and standard deviation (details in paper)..")
78
Use SNP array signals to infer CNV New Development Affymetrix SNP6 array: Genotype (SNP) and CNV probesets – two type of signals; don’t always follow the same distribution The auto selection of reference chromosome fails on cases with complex CNV patterns Modified algorithm: more flexible – Marker (instead of chrom.) based reference – Initial CNV inference, adjustment, final CNV inference
 and CNV probesets – two type of signals; don’t always follow the same distribution The auto selection of reference chromosome fails on cases with complex CNV patterns Modified algorithm: more flexible – Marker (instead of chrom.) based reference – Initial CNV inference, adjustment, final CNV inference")
79
Use SNP array signals to infer CNV Modified algorithm: Work in progress … 1.Select all SNP markers with heterozygous calls (‘AB’) as reference markers; use the empirical distribution of these marker signals as the initial reference distribution 2.Map all the SNP marker signals into (0,1) by probability-integral transformation using the above distribution 3.Map the above transformed signals to a target distribution – take N(0,1), to produce initially reference-aligned signals 4.Map the CNV marker signals to have the same distribution as the SNP markers via a quantile transformation 5.Perform initial CNV segmentation – windowed t test + run-length encoding 6.Assess each autosome and each “large” (>20 SNP markers) inferred CNV segments to identify problems 7.Correct the problems by adjusting the initial reference-aligned signals (step 3) chromosome by chromosome 8.Perform final CNV segmentation using corrected signals – windowed t test plus run-length encoding.
 as reference markers; use the empirical distribution of these marker signals as the initial reference distribution 2.Map all the SNP marker signals into (0,1) by probability-integral transformation using the above distribution 3.Map the above transformed signals to a target distribution – take N(0,1), to produce initially reference-aligned signals 4.Map the CNV marker signals to have the same distribution as the SNP markers via a quantile transformation 5.Perform initial CNV segmentation – windowed t test + run-length encoding 6.Assess each autosome and each large (>20 SNP markers) inferred CNV segments to identify problems 7.Correct the problems by adjusting the initial reference-aligned signals (step 3) chromosome by chromosome 8.Perform final CNV segmentation using corrected signals – windowed t test plus run-length encoding.")
80
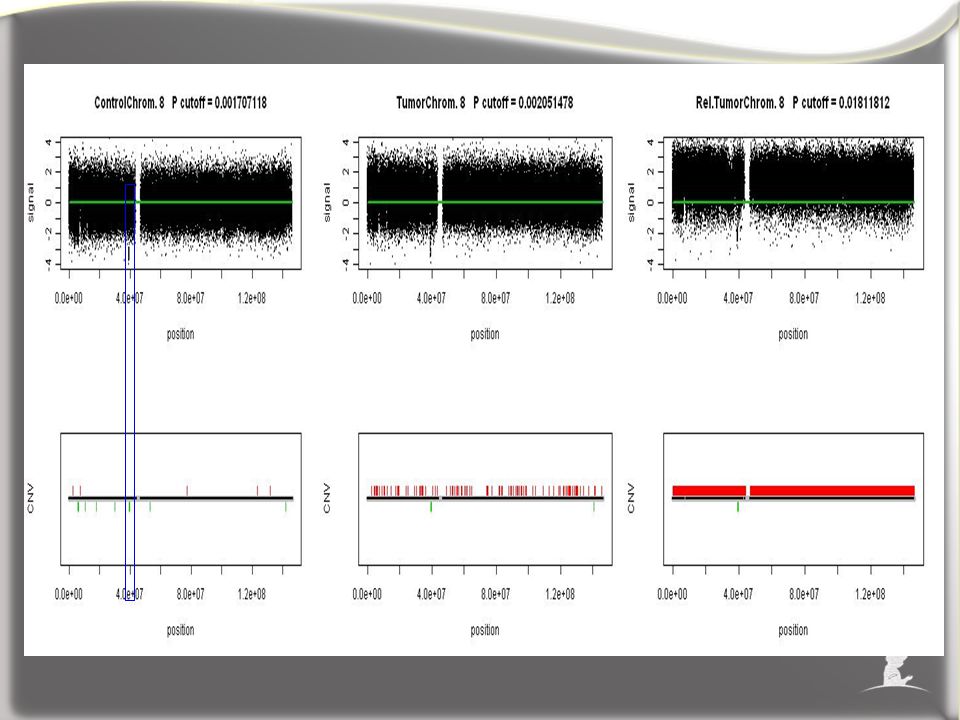
Male, Hypodiploid (<46 chromosomes) ALL and germline samples: Steps 3, 4, 5, 6 initial reference Tumor Germline overall 2cp, initially inferred loss t(9) Initial reference
 ALL and germline samples: Steps 3, 4, 5, 6 initial reference Tumor Germline overall 2cp, initially inferred loss t(9) Initial reference")
81
Use SNP array signals to infer CNV New Development Examples: – Two ALL cases -- one hypodiploid (<46 chr’s) with matched germline, one hyperdiploid (triploid, 66 chr’s) with matched germline and relapse samples
 with matched germline, one hyperdiploid (triploid, 66 chr’s) with matched germline and relapse samples")
82
Male, Hypodiploid ALL and germline samples Probeset signal: mean of probes, directly from the.cell file

83
Male, Hypodip ALL and matched germline %Het=26% %Het=0.16% %Het=29%

84
Germline Tumor

85
Male, Hypodiploid ALL and germline samples initial reference Tumor Germline overall 2cp, initially inferred loss t(9) Initial reference
 Initial reference")
86
Male, Triploid ALL, DNA index 1.46 67,XXY,-3,+8,-9,-16,-17,+20/66,idem,del(X)(p22.1),-8,del(10)(q22q26),-20,+mar 1-2(3)3(2)4-7(3)8(4)9(2)10-15(3)16-17(2)18-19(3)20(4)21-22(3)X(2) 6 2 12 4 2 18 4 6 4 6 2 total=66 +Y = 67 Matched germline sample, relapse sample Probeset signal: generated by Affy MAS 5.0 (?) package
(p22.1),-8,del(10)(q22q26),-20,+mar 1-2(3)3(2)4-7(3)8(4)9(2)10-15(3)16-17(2)18-19(3)20(4)21-22(3)X(2) total=66 +Y = 67 Matched germline sample, relapse sample Probeset signal: generated by Affy MAS 5.0 ( ) package")
87
67,XXY,-3,+8,-9,-16,-17,+20/66,idem,del(X)(p22.1),-8,del(10)(q22q26),-20,+mar 1-2(3)3(2)4-7(3)8(4)9(2)10-15(3)16-17(2)18-19(3)20(4)21-22(3)X(2) 6 2 12 4 2 18 4 6 4 6 2 total=66 +Y = 67
(p22.1),-8,del(10)(q22q26),-20,+mar 1-2(3)3(2)4-7(3)8(4)9(2)10-15(3)16-17(2)18-19(3)20(4)21-22(3)X(2) total=66 +Y = 67")
89
initial reference Relapsed Tumor Germline overall 2cp, initially inferred loss t(9) Initial reference
 Initial reference")
90
Use SNP array signals to infer CNV New Development Extension to NextGen sequence data – Use sequence coverage counts as raw signals – Preprocessing: Adjust signals for reference genome and sequence features that affect the depth of coverage (“mapability”, GC content, etc.) – The new alignment and segmentation algorithms consist of only single-marker based computation, thus straightforward to implement divide-and- conquer strategies and parallelization on multiprocessor or CPU cluster systems
 – The new alignment and segmentation algorithms consist of only single-marker based computation, thus straightforward to implement divide-and- conquer strategies and parallelization on multiprocessor or CPU cluster systems")
91
ACKNOWLEDGEMENTS NIH/NIGMS Pharmacogenetics Research Network and Database (U01 GM61393, U01GM61374, http://pharmgkb.org/) National Institutes of Health Cancer Center Support Grant P30 CA-21765, NIH The American Lebanese and Syrian Associated Charities (ALSAC). Stan Pounds, Deqing Pei, Xueyuan Cao – Biostatistics Mary Relling, William Evans, Jun Yang, Wenjian Yang – Pharmaceutical Science Ching-Hon Pui, Dario Campana – Oncology & Pathology Charles Mullighan, James Downing -- Pathology Geoff Neale, Yiping Fan -- Bioinformatics Javier Rojo – My first and most favorite Math Statistics teacher

92
THANK YOU !!

Similar presentations




















>")
