Download presentation
Presentation is loading. Please wait.

2
Computational Chemistry G. H. CHEN Department of Chemistry University of Hong Kong

3
In 1929, Dirac declared, “The underlying physical laws necessary for the mathematical theory of...the whole of chemistry are thus completely know, and the difficulty is only that the exact application of these laws leads to equations much too complicated to be soluble.” Beginning of Computational Chemistry Dirac

4
Computational Chemistry Quantum Chemistry Molecular Mechanics Bioinformatics Create & Analyse Bio-information Schr Ö dinger Equation F = M a

5
Mulliken,1966Fukui, 1981Hoffmann, 1981 Pople, 1998Kohn, 1998 Nobel Prizes for Computational Chemsitry

6
Computational Chemistry Industry CompanySoftware Gaussian Inc.Gaussian 94, Gaussian 98 Schrödinger Inc.Jaguar WavefunctionSpartanQ-Chem AccelrysInsightII, Cerius 2 HyperCubeHyperChem Celera Genomics (Dr. Craig Venter, formal Prof., SUNY, Baffalo; 98-01) Applications: material discovery, drug design & research R&D in Chemical & Pharmaceutical industries in 2000: US$ 80 billion Bioinformatics: Total Sales in 2001 US$ 225 million Project Sales in 2006US$ 1.7 billion
 Applications: material discovery, drug design & research R&D in Chemical & Pharmaceutical industries in 2000: US$ 80 billion Bioinformatics: Total Sales in 2001 US$ 225 million Project Sales in 2006US$ 1.7 billion.")
7
LODESTAR v1.02 --Localized Density Matrix: STAR performer http://yangtze.hku.hk Software Development at HKU

8
Quantum Chemistry Methods Ab initio molecular orbital methods Semiempirical molecular orbital methods Density functional method

9
H E Schr Ö dinger Equation Hamiltonian H = ( h 2 /2m h 2 /2m e ) i i 2 + Z Z e r i e 2 /r i i j e 2 /r ij Wavefunction Energy
 i i 2 + Z Z e r i e 2 /r i i j e 2 /r ij Wavefunction Energy")
10
Vitamin C C60 Cytochrome c heme OH + D 2 --> HOD + D energy

11
C 60 and Superconductor Applications: Magnet, Magnetic train, Power transportation What is superconductor? Electrical Current flows for ever !

12
Crystal Structure of C 60 solid Crystal Structure of K 3 C 60

13
K 3 C 60 is a Superconductor (T c = 19K) Erwin & Pickett, Science, 1991 GH Chen, Ph.D. Thesis, Caltech (1992) Vibration Spectrum of K 3 C 60 Effective Attraction ! The mechanism of superconductivity in K 3 C 60 was discovered using com- putational chemistry methods Varma et. al., 1991; Schluter et. al., 1992; Dresselhaus et. al., 1992;Chen & Goddard, 1992
 Vibration Spectrum of K 3 C 60 Effective Attraction . The mechanism of superconductivity in K 3 C 60 was discovered using com- putational chemistry methods Varma et. al., 1991; Schluter et. al., 1992; Dresselhaus et. al., 1992;Chen & Goddard,")
14
Carbon Nanotubes (Ijima, 1991)
")
15
STM Image of Carbon Nanotubes (Wildoer et. al., 1998) Calculated STM Image of a Carbon Nanotube (Rubio, 1999)
 Calculated STM Image of a Carbon Nanotube (Rubio, 1999).")
16
Computer Simulations (Saito, Dresselhaus, Louie et. al., 1992) Carbon Nanotubes (n,m): Conductor, if n-m = 3I I=0,±1,±2,±3,…;or Semiconductor, if n-m 3I Metallic Carbon Nanotubes: Conducting Wires Semiconducting Nanotubes:Transistors Molecular-scale circuits !1 nm transistor! 0.13 µm transistor! 30 nm transistor!
 Carbon Nanotubes (n,m): Conductor, if n-m = 3I I=0,±1,±2,±3,…;or Semiconductor, if n-m 3I Metallic Carbon Nanotubes: Conducting Wires Semiconducting Nanotubes:Transistors Molecular-scale circuits !1 nm transistor µm transistor. 30 nm transistor!.")
17
Wildoer, Venema, Rinzler, Smalley, Dekker, Nature 391, 59 (1998) Experimental Confirmations: Lieber et. al. 1993; Dravid et. al., 1993; Iijima et. al. 1993; Smalley et. al. 1998; Haddon et. al. 1998; Liu et. al. 1999

18
Science 9 th November, 2001 Logic gates (and circuits) with carbon nanotuce transistor Science 7 th July, 2000 Carbon nanotube-Based nonvolatile RAM for molecular computing
 with carbon nanotuce transistor Science 7 th July, 2000 Carbon nanotube-Based nonvolatile RAM for molecular computing")
20
Nanoelectromechanical Systems (NEMS) K.E. Drexler, Nanosystems: Molecular Machinery, Manufacturing and Computation (Wiley, New York, 1992).
..")
21
Large Gear Drives Small Gear G. Hong et. al., 1999

22
Nano-oscillators Zhao, Ma, Chen & Jiang, Phys. Rev. Lett. 2003 Nanoscopic Electromechanical Device (NEMS)
.")
23
Hibernation Awakening Oscillation

24
Quantum mechanical investigation of the field emission from the tips of carbon nanotubes Zettl, PRL 2001 Zheng, Chen, Li, Deng & Xu, Phys. Rev. Lett. 2004

25
Computer-Aided Drug Design GENOMICS Human Genome Project

26
ALDOSE REDUCTASE Diabetes Diabetic Complications Glucose Sorbitol

27
Design of Aldose Reductase Inhibitors Aldose Reductase Inhibitor Hu, Chen & Chau, J. Mol. Graph. Mod. 24 (2006)
.")
28
Database for Functional Groups LogIC50: 0.6382,1.0 LogIC50: 0.6861,0.88 Prediction: Drug Leads Structure-activity-relation

30
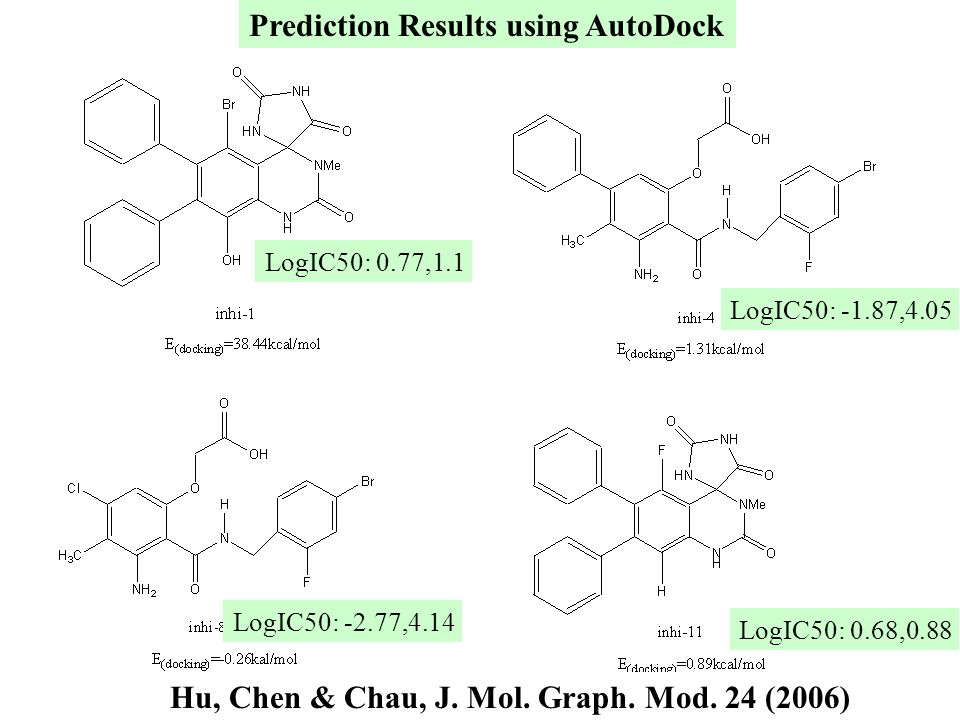
LogIC50: 0.77,1.1 LogIC50: -1.87,4.05 LogIC50: -2.77,4.14 LogIC50: 0.68,0.88 Prediction Results using AutoDock Hu, Chen & Chau, J. Mol. Graph. Mod. 24 (2006)
.")
31
Computer-aided drug design Chemical Synthesis Screening using in vitro assay Animal Tests Clinical Trials

32
Bioinformatics Improve content & utility of bio-databases Develop tools for data generation, capture & annotation Develop tools for comprehensive functional studies Develop tools for representing & analyzing sequence similarity & variation

35
Computational Chemistry Increasingly important field in chemistry Help to understand experimental results Provide guidelines to experimentists Application in Materials & Pharmaceutical industries Future: simulate nano-size materials, bulk materials; replace experimental R&D Objective: More and more research & development to be performed on computers and Internet instead in the laboratories

36
Quantum Chemistry G. H. Chen Department of Chemistry University of Hong Kong

37
Contributors: Hartree, Fock, Slater, Hund, Mulliken, Lennard-Jones, Heitler, London, Brillouin, Koopmans, Pople, Kohn Application: Chemistry, Condensed Matter Physics, Molecular Biology, Materials Science, Drug Discovery

38
Emphasis Hartree-Fock method Concepts Hands-on experience Text Book “Quantum Chemistry”, 4th Ed. Ira N. Levine http://yangtze.hku.hk/lecture/chem3504-3.ppt

39
Contents 1. Variation Method 2. Hartree-Fock Self-Consistent Field Method 3. Perturbation Theory 4. Semiempirical Methods

40
The Variation Method Consider a system whose Hamiltonian operator H is time independent and whose lowest-energy eigenvalue is E 1. If is any normalized, well- behaved function that satisfies the boundary conditions of the problem, then * H d E 1 The variation theorem

41
Proof: Expand ii n the basis set { k } = k k k where { k } are coefficients H k = E k k then * H dd k j k * j E j kj = k | k | 2 E k E 1 k | k | 2 = E 1 Since is normalized, * dd k | k | 2 = 1

42
i. : trial function is used to evaluate the upper limit of ground state energy E 1 ii. = ground state wave function, * H d E 1 iii. optimize paramemters in by minimizing * H d * d

43
Requirements for the trial wave function: i. zero at boundary; ii. smoothness a maximum in the center. Trial wave function: = x (l - x) Application to a particle in a box of infinite depth 0 l
 Application to a particle in a box of infinite depth 0 l.")
44
* H dx = -(h 2 /8 2 m) (lx-x 2 ) d 2 (lx-x 2 )/dx 2 dx = h 2 /(4 2 m) (x 2 - lx) dx = h 2 l 3 /(24 2 m) * dx = x 2 (l-x) 2 dx = l 5 /30 E = 5h 2 /(4 2 l 2 m) h 2 /(8ml 2 ) = E 1
 (lx-x 2 ) d 2 (lx-x 2 )/dx 2 dx = h 2 /(4 2 m) (x 2 - lx) dx = h 2 l 3 /(24 2 m) * dx = x 2 (l-x) 2 dx = l 5 /30 E = 5h 2 /(4 2 l 2 m) h 2 /(8ml 2 ) = E 1")
45
(1) Construct a wave function (c 1,c 2, ,c m ) (2) Calculate the energy of : E E (c 1,c 2, ,c m ) (3) Choose {c j * } (i=1,2, ,m) so that E is minimum Variational Method
 Construct a wave function (c 1,c 2, ,c m ) (2) Calculate the energy of : E E (c 1,c 2, ,c m ) (3) Choose {c j * } (i=1,2, ,m) so that E is minimum Variational Method")
46
Example: one-dimensional harmonic oscillator Potential: V(x) = (1/2) kx 2 = (1/2) m 2 x 2 = 2 2 m 2 x 2 Trial wave function for the ground state: (x) = exp(-cx 2 ) * H dx = -(h 2 /8 2 m) exp(-cx 2 ) d 2 [exp(-cx 2 )]/dx 2 dx + 2 2 m 2 x 2 exp(-2cx 2 ) dx = (h 2 /4 2 m) ( c/8) 1/2 + 2 m 2 ( /8c 3 ) 1/2 * dx = exp(-2cx 2 ) dx = ( /2) 1/2 c -1/2 E = W = (h 2 /8 2 m)c + ( 2 /2)m 2 /c
![Example: one-dimensional harmonic oscillator Potential: V(x) = (1/2) kx 2 = (1/2) m 2 x 2 = 2 2 m 2 x 2 Trial wave function for the ground state: (x) = exp(-cx 2 ) * H dx = -(h 2 /8 2 m) exp(-cx 2 ) d 2 [exp(-cx 2 )]/dx 2 dx + 2 2 m 2 x 2 exp(-2cx 2 ) dx = (h 2 /4 2 m) ( c/8) 1/2 + 2 m 2 ( /8c 3 ) 1/2 * dx = exp(-2cx 2 ) dx = ( /2) 1/2 c -1/2 E = W = (h 2 /8 2 m)c + ( 2 /2)m 2 /c](http://images.slideplayer.com/16/5051037/slides/slide_46.jpg "Example: one-dimensional harmonic oscillator Potential: V(x) = (1/2) kx 2 = (1/2) m 2 x 2 = 2 2 m 2 x 2 Trial wave function for the ground state: (x) = exp(-cx 2 ) * H dx = -(h 2 /8 2 m) exp(-cx 2 ) d 2 [exp(-cx 2 )]/dx 2 dx + 2 2 m 2 x 2 exp(-2cx 2 ) dx = (h 2 /4 2 m) ( c/8) 1/2 + 2 m 2 ( /8c 3 ) 1/2 * dx = exp(-2cx 2 ) dx = ( /2) 1/2 c -1/2 E = W = (h 2 /8 2 m)c + ( 2 /2)m 2 /c")
47
To minimize W, 0 = dW/dc = h 2 /8 2 m - ( 2 /2)m 2 c -2 c = 2 2 m/h W = (1/2) h
m 2 c -2 c = 2 2 m/h W = (1/2) h")
48
. E 3 3 E 2 2 E 1 1 Extension of Variation Method For a wave function which is orthogonal to the ground state wave function 1, i.e. d * 1 = 0 E = d * H / d * > E 2 the first excited state energy

49
The trial wave function : d * 1 = 0 k=1 a k k d * 1 = |a 1 | 2 = 0 E = d * H / d * = k=2 |a k | 2 E k / k=2 |a k | 2 > k=2 |a k | 2 E 2 / k=2 |a k | 2 = E 2

50
e + + 1 2 c 1 1 + c 2 2 W = H d d = (c 1 2 H 11 + 2c 1 c 2 H 12 + c 2 2 H 22 ) / (c 1 2 + 2c 1 c 2 S + c 2 2 ) W (c 1 2 + 2c 1 c 2 S + c 2 2 ) = c 1 2 H 11 + 2c 1 c 2 H 12 + c 2 2 H 22 Application to H 2 +
 / (c c 1 c 2 S + c 2 2 ) W (c c 1 c 2 S + c 2 2 ) = c 1 2 H c 1 c 2 H 12 + c 2 2 H 22 Application to H 2 +")
51
Partial derivative with respect to c 1 ( W/ c 1 = 0) : W (c 1 + S c 2 ) = c 1 H 11 + c 2 H 12 Partial derivative with respect to c 2 ( W/ c 2 = 0) : W (S c 1 + c 2 ) = c 1 H 12 + c 2 H 22 (H 11 - W) c 1 + (H 12 - S W) c 2 = 0 (H 12 - S W) c 1 + (H 22 - W) c 2 = 0
 : W (c 1 + S c 2 ) = c 1 H 11 + c 2 H 12 Partial derivative with respect to c 2 ( W/ c 2 = 0) : W (S c 1 + c 2 ) = c 1 H 12 + c 2 H 22 (H 11 - W) c 1 + (H 12 - S W) c 2 = 0 (H 12 - S W) c 1 + (H 22 - W) c 2 = 0")
52
To have nontrivial solution: H 11 - WH 12 - S W H 12 - S WH 22 - W For H 2 +, H 11 = H 22 ; H 12 < 0. Ground State: E g = W 1 = (H 11 +H 12 ) / (1+S) = ( ) / 2(1+S) 1/2 Excited State: E e = W 2 = (H 11 -H 12 ) / (1-S) = ( ) / 2(1-S) 1/2 = 0 bonding orbital Anti-bonding orbital
 / (1+S) = ( ) / 2(1+S) 1/2 Excited State: E e = W 2 = (H 11 -H 12 ) / (1-S) = ( ) / 2(1-S) 1/2 = 0 bonding orbital Anti-bonding orbital.")
53
Results: D e = 1.76 eV, R e = 1.32 A Exact: D e = 2.79 eV, R e = 1.06 A 1 eV = 23.0605 kcal / mol

54
Trial wave function: k 3/2 -1/2 exp(-kr) E g = W 1 (k,R) at each R, choose k so that W 1 / k = 0 Results: D e = 2.36 eV, R e = 1.06 A Resutls: D e = 2.73 eV, R e = 1.06 A 1s1s 2p2p Inclusion of other atomic orbitals Further Improvements H -1/2 exp(-r) He + 2 3/2 -1/2 exp(-2r) Optimization of 1s orbitals
 E g = W 1 (k,R) at each R, choose k so that W 1 / k = 0 Results: D e = 2.36 eV, R e = 1.06 A Resutls: D e = 2.73 eV, R e = 1.06 A 1s1s 2p2p Inclusion of other atomic orbitals Further Improvements H -1/2 exp(-r) He + 2 3/2 -1/2 exp(-2r) Optimization of 1s orbitals")
55
a 11 x 1 + a 12 x 2 = b 1 a 21 x 1 + a 22 x 2 = b 2 (a 11 a 22 -a 12 a 21 ) x 1 = b 1 a 22 -b 2 a 12 (a 11 a 22 -a 12 a 21 ) x 2 = b 2 a 11 -b 1 a 21 Linear Equations 1. two linear equations for two unknown, x 1 and x 2

56
Introducing determinant: a 11 a 12 = a 11 a 22 -a 12 a 21 a 21 a 22 a 11 a 12 b 1 a 12 x 1 = a 21 a 22 b 2 a 22 a 11 a 12 a 11 b 1 x 2 = a 21 a 22 a 21 b 2

57
Our case: b 1 = b 2 = 0, homogeneous 1. trivial solution: x 1 = x 2 = 0 2. nontrivial solution: a 11 a 12 = 0 a 21 a 22 n linear equations for n unknown variables a 11 x 1 + a 12 x 2 +... + a 1n x n = b 1 a 21 x 1 + a 22 x 2 +... + a 2n x n = b 2............................................ a n1 x 1 + a n2 x 2 +... + a nn x n = b n

58
a 11 a 12... a 1,k-1 b 1 a 1,k+1... a 1n a 21 a 22... a 2,k-1 b 2 a 2,k+1... a 2n det(a ij ) x k =............ a n1 a n2... a n,k-1 b 2 a n,k+1... a nn where, a 11 a 12...a 1n a 21 a 22...a 2n det(a ij ) =...... a n1 a n2...a nn
 x k = a n1 a n2... a n,k-1 b 2 a n,k+1... a nn where, a 11 a 12...a 1n a 21 a 22...a 2n det(a ij ) = a n1 a n2...a nn.")
59
a 11 a 12...a 1,k-1 b 1 a 1,k+1...a 1n a 21 a 22...a 2,k-1 b 2 a 2,k+1...a 2n............ a n1 a n2...a n,k-1 b 2 a n,k+1...a nn x k = det(a ij ) inhomogeneous case: b k = 0 for at least one k
 inhomogeneous case: b k = 0 for at least one k.")
60
(a) travial case: x k = 0, k = 1, 2,..., n (b) nontravial case: det(a ij ) = 0 homogeneous case: b k = 0, k = 1, 2,..., n For a n-th order determinant, n det(a ij ) = a lk C lk l=1 where, C lk is called cofactor
 travial case: x k = 0, k = 1, 2,..., n (b) nontravial case: det(a ij ) = 0 homogeneous case: b k = 0, k = 1, 2,..., n For a n-th order determinant, n det(a ij ) = a lk C lk l=1 where, C lk is called cofactor")
61
Trial wave function is a variation function which is a combination of n linear independent functions { f 1, f 2,... f n }, c 1 f 1 + c 2 f 2 +... + c n f n n [( H ik - S ik W ) c k ] = 0 i=1,2,...,n k=1 S ik d f i f k H ik d f i H f k W d H d
 c k ] = 0 i=1,2,...,n k=1 S ik d f i f k H ik d f i H f k W d H d .")
62
(i) W 1 W 2 ... W n are n roots of Eq.(1), (ii) E 1 E 2 ... E n E n+1 ... are energies of eigenstates; then, W 1 E 1, W 2 E 2,..., W n E n Linear variational theorem

63
Molecular Orbital (MO): = c 1 1 + c 2 2 ( H 11 - W ) c 1 + ( H 12 - SW ) c 2 = 0 S 11 =1 ( H 21 - SW ) c 1 + ( H 22 - W ) c 2 = 0 S 22 =1 Generally : i a set of atomic orbitals, basis set LCAO-MO = c 1 1 + c 2 2 +...... + c n n linear combination of atomic orbitals n ( H ik - S ik W ) c k = 0 i = 1, 2,......, n k=1 H ik d i * H k S ik d i * k S kk = 1
 c k = 0 i = 1, 2,......, n k=1 H ik d i * H k S ik d i * k S kk = 1.")
64
Hamiltonian H = ( h 2 /2m h 2 /2m e ) i i 2 + Z Z e r i e 2 /r i i j e 2 /r ij H r i ;r r i ;r The Born-Oppenheimer Approximation
 i i 2 + Z Z e r i e 2 /r i i j e 2 /r ij H r i ;r r i ;r The Born-Oppenheimer Approximation")
65
r i ;r el r i ;r N r el ( r ) = h 2 /2m e ) i i 2 i e 2 /r i i j e 2 /r ij V NN = Z Z e r H el ( r ) el r i ;r el ( r ) el r i ;r (3) H N = ( h 2 /2m U( r ) U( r ) = el ( r ) + V NN H N ( r ) N r N r The Born-Oppenheimer Approximation:
 = h 2 /2m e ) i i 2 i e 2 /r i i j e 2 /r ij V NN = Z Z e r H el ( r ) el r i ;r el ( r ) el r i ;r (3) H N = ( h 2 /2m U( r ) U( r ) = el ( r ) + V NN H N ( r ) N r N r The Born-Oppenheimer Approximation:")
66
Assignment Calculate the ground state energy and bond length of H 2 using the HyperChem with the 6-31G (Hint: Born-Oppenheimer Approximation)
")
67
e + + e two electrons cannot be in the same state. Hydrogen Molecule H 2 The Pauli principle

68
Since two wave functions that correspond to the same state can differ at most by a constant factor = c 2 a b c 1 a b =c 2 a b +c 2 c 1 a b c 1 = c 2 c 2 c 1 = 1 Therefore: c 1 = c 2 = 1 According to the Pauli principle, c 1 = c 2 = 1 Wave function: = a b + c 1 a b = a b + c 1 a b

69
Wave function f H 2 : ! [ ! The Pauli principle (different version) the wave function of a system of electrons must be antisymmetric with respect to interchanging of any two electrons. Slater Determinant
 the wave function of a system of electrons must be antisymmetric with respect to interchanging of any two electrons. Slater Determinant.")
70
E =2 d T e +V eN ) + V NN + d d e 2 /r 12 | = i=1,2 f ii + J 12 + V NN To minimize E under the constraint d | use Lagrange’s method: L = E d L = E d 4 d T e +V eN ) +4 d d e 2 /r 12 d Energy: E
 + V NN + d d e 2 /r 12 | = i=1,2 f ii + J 12 + V NN To minimize E under the constraint d | use Lagrange’s method: L = E d L = E d 4 d T e +V eN ) +4 d d e 2 /r 12 d Energy: E ")
71
[ T e +V eN + d e 2 /r 12 f + J f(1) = T e (1)+V eN (1) one electron operator J(1) = d e 2 /r 12 two electron Coulomb operator Average Hamiltonian Hartree-Fock equation
 = T e (1)+V eN (1) one electron operator J(1) = d e 2 /r 12 two electron Coulomb operator Average Hamiltonian Hartree-Fock equation")
72
f(1) is the Hamiltonian of electron 1 in the absence of electron 2; J(1) is the mean Coulomb repulsion exerted on electron 1 by 2; is the energy of orbital LCAO-MO: c 1 1 + c 2 2 Multiple 1 from the left and then integrate : c 1 F 11 + c 2 F 12 = (c 1 + S c 2 )
 is the Hamiltonian of electron 1 in the absence of electron 2; J(1) is the mean Coulomb repulsion exerted on electron 1 by 2; is the energy of orbital LCAO-MO: c 1 1 + c 2 2 Multiple 1 from the left and then integrate : c 1 F 11 + c 2 F 12 = (c 1 + S c 2 )")
73
Multiple 2 from the left and then integrate : c 1 F 12 + c 2 F 22 = (S c 1 + c 2 ) where, F ij = d i * ( f + J ) j = H ij + d i * J j S = d 1 2 (F 11 - ) c 1 + (F 12 - S ) c 2 = 0 (F 12 - S ) c 1 + (F 22 - ) c 2 = 0
 where, F ij = d i * ( f + J ) j = H ij + d i * J j S = d 1 2 (F 11 - ) c 1 + (F 12 - S ) c 2 = 0 (F 12 - S ) c 1 + (F 22 - ) c 2 = 0")
74
Secular Equation: F 11 - F 12 - S F 12 - S F 22 - bonding orbital: 1 = (F 11 +F 12 ) / (1+S) = ( ) / 2(1+S) 1/2 antibonding orbital: 2 = (F 11 -F 12 ) / (1-S ) = ( ) / 2(1-S) 1/2
 / (1+S) = ( ) / 2(1+S) 1/2 antibonding orbital: 2 = (F 11 -F 12 ) / (1-S ) = ( ) / 2(1-S) 1/2")
75
Molecular Orbital Configurations of Homo nuclear Diatomic Molecules H 2, Li 2, O, He 2, etc Moecule Bond order De/eV H 2 + 2.79 H 2 1 4.75 He 2 + 1.08 He 2 0 0.0009 Li 2 1 1.07 Be 2 0 0.10 C 2 2 6.3 N 2 + 8.85 N 2 3 9.91 O 2 + 2 6.78 O 2 2 5.21 The more the Bond Order is, the stronger the chemical bond is.

76
Bond Order: one-half the difference between the number of bonding and antibonding electrons

77
-------- -------- 1 -------- -------- 2 1 2 1 2 = 1/ 2 [ 1 2 2 1

78
d d H d d (T 1 +V 1N +T 2 +V 2N +V 12 +V NN ) 1 T 1 +V 1N | 1 2 T 2 +V 2N | 2 + 1 2 V 12 1 2 1 2 V 12 1 2 + V NN = i i T 1 +V 1N | i + 1 2 V 12 1 2 1 2 V 12 1 2 + V NN = i=1,2 f ii + J 12 K 12 + V NN
 1 T 1 +V 1N | 1 2 T 2 +V 2N | 2 + 1 2 V 12 1 2 1 2 V 12 1 2 + V NN = i i T 1 +V 1N | i + 1 2 V 12 1 2 1 2 V 12 1 2 + V NN = i=1,2 f ii + J 12 K 12 + V NN")
79
Particle One: f(1) + J 2 (1) K 2 (1) Particle Two: f(2) + J 1 (2) K 1 (2) f(j) h 2 /2m e ) j 2 Z r j J j (1) dr j * e 2 /r 12 j K j (1) j dr j * e 2 /r 12 Average Hamiltonian
 + J 2 (1) K 2 (1) Particle Two: f(2) + J 1 (2) K 1 (2) f(j) h 2 /2m e ) j 2 Z r j J j (1) dr j * e 2 /r 12 j K j (1) j dr j * e 2 /r 12 Average Hamiltonian")
80
f(1)+ J 2 (1) K 2 (1) 1 (1) 1 1 (1) f(2)+ J 1 (2) K 1 (2) 2 (2) 2 2 (2) F(1) f(1)+ J 2 (1) K 2 (1) Fock operator for 1 F(2) f(2)+ J 1 (2) K 1 (2) Fock operator for 2 Hartree-Fock Equation: Fock Operator:
+ J 2 (1) K 2 (1) 1 (1) 1 1 (1) f(2)+ J 1 (2) K 1 (2) 2 (2) 2 2 (2) F(1) f(1)+ J 2 (1) K 2 (1) Fock operator for 1 F(2) f(2)+ J 1 (2) K 1 (2) Fock operator for 2 Hartree-Fock Equation: Fock Operator:")
81
1.At the Hartree-Fock Level there are two possible Coulomb integrals contributing the energy between two electrons i and j: Coulomb integrals J ij and exchange integral K ij ; 2.For two electrons with different spins, there is only Coulomb integral J ij ; 3. For two electrons with the same spins, both Coulomb and exchange integrals exist. Summary

82
4.Total Hartree-Fock energy consists of the contributions from one-electron integrals f ii and two-electron Coulomb integrals J ij and exchange integrals K ij ; 5.At the Hartree-Fock Level there are two possible Coulomb potentials (or operators) between two electrons i and j: Coulomb operator and exchange operator; J j (i) is the Coulomb potential (operator) that i feels from j, and K j (i) is the exchange potential (operator) that that i feels from j.
 between two electrons i and j: Coulomb operator and exchange operator; J j (i) is the Coulomb potential (operator) that i feels from j, and K j (i) is the exchange potential (operator) that that i feels from j.")
83
6. Fock operator (or, average Hamiltonian) consists of one-electron operators f(i) and Coulomb operators J j (i) and exchange operators K j (i)
 consists of one-electron operators f(i) and Coulomb operators J j (i) and exchange operators K j (i) .")
84
N electrons spin up and N electrons spin down. Fock matrix for an electron 1 with spin up: F (1 ) = f (1 ) + j [ J j (1 ) K j (1 ) ] + j J j (1 ) j=1 ,N j=1 ,N Fock matrix for an electron 1 with spin down: F (1 ) = f (1 ) + j [ J j (1 ) K j (1 ) ] + j J j (1 ) j=1 ,N j=1 ,N
 = f (1 ) + j [ J j (1 ) K j (1 ) ] + j J j (1 ) j=1 ,N j=1 ,N Fock matrix for an electron 1 with spin down: F (1 ) = f (1 ) + j [ J j (1 ) K j (1 ) ] + j J j (1 ) j=1 ,N j=1 ,N .")
85
f(1) h 2 /2m e ) 1 2 N Z N r 1N J j (1) dr j e 2 /r 12 j K j (1) j dr j * e 2 /r 12 Energy = j f jj + j f jj +(1/2) i j ( J ij K ij ) + (1/2) i j ( J ij K ij ) + i j J ij + V NN i=1,N j=1,N
 h 2 /2m e ) 1 2 N Z N r 1N J j (1) dr j e 2 /r 12 j K j (1) j dr j * e 2 /r 12 Energy = j f jj + j f jj +(1/2) i j ( J ij K ij ) + (1/2) i j ( J ij K ij ) + i j J ij + V NN i=1,N j=1,N ")
86
f jj f jj j f j J ij J ij j (2) J i j (2) K ij K ij j (2) K i j (2) J ij J ij j (2) J i j (2) F(1) = f (1) + j=1,n/2 [ 2J j (1) K j (1) ] Energy = 2 j=1,n/2 f jj + i=1,n/2 j=1,n/2 ( 2J ij K ij ) + V NN Close subshell case: ( N = N = n/2 )
![f jj f jj j f j J ij J ij j (2) J i j (2) K ij K ij j (2) K i j (2) J ij J ij j (2) J i j (2) F(1) = f (1) + j=1,n/2 [ 2J j (1) K j (1) ] Energy = 2 j=1,n/2 f jj + i=1,n/2 j=1,n/2 ( 2J ij K ij ) + V NN Close subshell case: ( N = N = n/2 )](http://images.slideplayer.com/16/5051037/slides/slide_86.jpg "f jj f jj j f j J ij J ij j (2) J i j (2) K ij K ij j (2) K i j (2) J ij J ij j (2) J i j (2) F(1) = f (1) + j=1,n/2 [ 2J j (1) K j (1) ] Energy = 2 j=1,n/2 f jj + i=1,n/2 j=1,n/2 ( 2J ij K ij ) + V NN Close subshell case: ( N = N = n/2 )")
87
1. Many-Body Wave Function is approximated by Slater Determinant 2. Hartree-Fock Equation F i = i i F Fock operator i the i-th Hartree-Fock orbital i the energy of the i-th Hartree-Fock orbital Hartree-Fock Method

88
3. Roothaan Method (introduction of Basis functions) i = k c ki k LCAO-MO { k } is a set of atomic orbitals (or basis functions) 4. Hartree-Fock-Roothaan equation j ( F ij - i S ij ) c ji = 0 F ij i F j S ij i j 5. Solve the Hartree-Fock-Roothaan equation self-consistently
 i = k c ki k LCAO-MO { k } is a set of atomic orbitals (or basis functions) 4. Hartree-Fock-Roothaan equation j ( F ij - i S ij ) c ji = 0 F ij i F j S ij i j 5. Solve the Hartree-Fock-Roothaan equation self-consistently.")
89
a b c d n f(1) e f g h n a f(1) e b c d n f g h n = a f(1) e if b=f, c=g,..., d=h; 0, otherwise a b c d n V 12 | e f g h n a b V 12 e f c d n g h n = a b V 12 e f if c=g,..., d=h; 0, otherwise The Condon-Slater Rules
 e f g h n a f(1) e b c d n f g h n = a f(1) e if b=f, c=g,..., d=h; 0, otherwise a b c d n V 12 | e f g h n a b V 12 e f c d n g h n = a b V 12 e f if c=g,..., d=h; 0, otherwise The Condon-Slater Rules")
90
------- the lowest unoccupied molecular orbital ------- the highest occupied molecular orbital ------- ------- The energy required to remove an electron from a closed-shell atom or molecules is well approximated by minus the orbital energy of the AO or MO from which the electron is removed. HOMO LUMO Koopman’s Theorem

91
# HF/6-31G(d) Route section water energy Title 0 1 Molecule Specification O -0.464 0.177 0.0 (in Cartesian coordinates H -0.464 1.137 0.0 H 0.441 -0.143 0.0
 Route section water energy Title 0 1 Molecule Specification O (in Cartesian coordinates H H")
92
Slater-type orbitals (STO) nlm = N r n-1 exp( r/a 0 ) Y lm ( , ) the orbital exponent * is used instead of in the textbook Gaussian type functions g ijk = N x i y j z k exp(- r 2 ) (primitive Gaussian function) p = u d up g u (contracted Gaussian-type function, CGTF) u = {ijk}p = {nlm} Basis Set i = p c ip p
 nlm = N r n-1 exp( r/a 0 ) Y lm ( , ) the orbital exponent * is used instead of in the textbook Gaussian type functions g ijk = N x i y j z k exp(- r 2 ) (primitive Gaussian function) p = u d up g u (contracted Gaussian-type function, CGTF) u = {ijk}p = {nlm} Basis Set i = p c ip p")
93
Basis set of GTFs STO-3G, 3-21G, 4-31G, 6-31G, 6-31G*, 6-31G** ------------------------------------------------------------------------------------- complexity & accuracy Minimal basis set: one STO for each atomic orbital (AO) STO-3G: 3 GTFs for each atomic orbital 3-21G: 3 GTFs for each inner shell AO 2 CGTFs (w/ 2 & 1 GTFs) for each valence AO 6-31G: 6 GTFs for each inner shell AO 2 CGTFs (w/ 3 & 1 GTFs) for each valence AO 6-31G*: adds a set of d orbitals to atoms in 2nd & 3rd rows 6-31G**: adds a set of d orbitals to atoms in 2nd & 3rd rows and a set of p functions to hydrogen Polarization Function
 STO-3G: 3 GTFs for each atomic orbital 3-21G: 3 GTFs for each inner shell AO 2 CGTFs (w/ 2 & 1 GTFs) for each valence AO 6-31G: 6 GTFs for each inner shell AO 2 CGTFs (w/ 3 & 1 GTFs) for each valence AO 6-31G*: adds a set of d orbitals to atoms in 2nd & 3rd rows 6-31G**: adds a set of d orbitals to atoms in 2nd & 3rd rows and a set of p functions to hydrogen Polarization Function")
94
Diffuse Basis Sets: For excited states and in anions where electronic density is more spread out, additional basis functions are needed. Diffuse functions to 6-31G basis set as follows: 6-31G* - adds a set of diffuse s & p orbitals to atoms in 1st & 2nd rows (Li - Cl). 6-31G** - adds a set of diffuse s and p orbitals to atoms in 1st & 2nd rows (Li- Cl) and a set of diffuse s functions to H Diffuse functions + polarisation functions: 6-31+G*, 6-31++G*, 6-31+G** and 6-31++G** basis sets. Double-zeta (DZ) basis set: two STO for each AO
. 6-31G** - adds a set of diffuse s and p orbitals to atoms in 1st & 2nd rows (Li- Cl) and a set of diffuse s functions to H Diffuse functions + polarisation functions: 6-31+G*, G*, 6-31+G** and G** basis sets. Double-zeta (DZ) basis set: two STO for each AO.")
95
6-31G for a carbon atom:(10s4p) [3s2p] 1s2s2p i (i=x,y,z) 6GTFs 3GTFs 1GTF3GTFs 1GTF 1CGTF 1CGTF 1CGTF 1CGTF 1CGTF (s)(s) (s) (p) (p)
![6-31G for a carbon atom:(10s4p) [3s2p] 1s2s2p i (i=x,y,z) 6GTFs 3GTFs 1GTF3GTFs 1GTF 1CGTF 1CGTF 1CGTF 1CGTF 1CGTF (s)(s) (s) (p) (p)](http://images.slideplayer.com/16/5051037/slides/slide_95.jpg "6-31G for a carbon atom:(10s4p) [3s2p] 1s2s2p i (i=x,y,z) 6GTFs 3GTFs 1GTF3GTFs 1GTF 1CGTF 1CGTF 1CGTF 1CGTF 1CGTF (s)(s) (s) (p) (p)")
96
Minimal basis set: One STO for each inner-shell and valence-shell AO of each atom example: C 2 H 2 (2S1P/1S) C: 1S, 2S, 2P x,2P y,2P z H: 1S total 12 STOs as Basis set Double-Zeta (DZ) basis set: two STOs for each and valence-shell AO of each atom example: C 2 H 2 (4S2P/2S) C: two 1S, two 2S, two 2P x, two 2P y,two 2P z H: two 1S (STOs) total 24 STOs as Basis set
 C: 1S, 2S, 2P x,2P y,2P z H: 1S total 12 STOs as Basis set Double-Zeta (DZ) basis set: two STOs for each and valence-shell AO of each atom example: C 2 H 2 (4S2P/2S) C: two 1S, two 2S, two 2P x, two 2P y,two 2P z H: two 1S (STOs) total 24 STOs as Basis set")
97
Split -Valence (SV) basis set Two STOs for each inner-shell and valence-shell AO One STO for each inner-shell AO Double-zeta plus polarization set(DZ+P, or DZP) Additional STO w/l quantum number larger than the l max of the valence - shell ( 2P x, 2P y,2P z ) to H Five 3d Aos to Li - Ne, Na -Ar C 2 H 5 O Si H 3 : (6s4p1d/4s2p1d/2s1p) Si C,O H
 basis set Two STOs for each inner-shell and valence-shell AO One STO for each inner-shell AO Double-zeta plus polarization set(DZ+P, or DZP) Additional STO w/l quantum number larger than the l max of the valence - shell ( 2P x, 2P y,2P z ) to H Five 3d Aos to Li - Ne, Na -Ar C 2 H 5 O Si H 3 : (6s4p1d/4s2p1d/2s1p) Si C,O H")
98
Assignment: Calculate the structure, ground state energy, molecular orbital energies, and vibrational modes and frequencies of a water molecule using Hartree-Fock method with 3-21G basis set. (due 30/10)
.")
99
1. L-Click on (click on left button of Mouse) “Startup”, and select and L-Click on “Program/Hyperchem”. 2. Select “Build’’ and turn on “Explicit Hydrogens”. 3. Select “Display” and make sure that “Show Hydrogens” is on; L-Click on “Rendering” and double L-Click “Spheres”. 4. Double L-Click on “Draw” tool box and double L-Click on “O”. 5. Move the cursor to the workspace, and L-Click & release. 6. L-Click on “Magnify/Shrink” tool box, move the cursor to the workspace; L-press and move the cursor inward to reduce the size of oxygen atom. 7. Double L-Click on “Draw” tool box, and double L-Click on “H”; Move the cursor close to oxygen atom and L-Click & release. A hydrogen atom appears. Draw second hydrogen atom using the same procedure. Ab Initio Molecular Orbital Calculation: H 2 O (using HyperChem)
 Startup , and select and L-Click on Program/Hyperchem . 2. Select Build’’ and turn on Explicit Hydrogens . 3. Select Display and make sure that Show Hydrogens is on; L-Click on Rendering and double L-Click Spheres . 4. Double L-Click on Draw tool box and double L-Click on O . 5. Move the cursor to the workspace, and L-Click & release. 6. L-Click on Magnify/Shrink tool box, move the cursor to the workspace; L-press and move the cursor inward to reduce the size of oxygen atom. 7. Double L-Click on Draw tool box, and double L-Click on H ; Move the cursor close to oxygen atom and L-Click & release. A hydrogen atom appears. Draw second hydrogen atom using the same procedure. Ab Initio Molecular Orbital Calculation: H 2 O (using HyperChem).")
100
8. L-Click on “Setup” & select “Ab Initio”; double L-Click on 3-21G; then L-Click on “Option”, select “UHF”, and set “Charge” to 0 and “Multiplicity” to 1. 9. L-Click “Compute”, and select “Geometry Optimization”, and L-Click on “OK”; repeat the step till “Conv=YES” appears in the bottom bar. Record the energy. 10.L-Click “Compute” and L-Click “Orbitals”; select a energy level, record the energy of each molecular orbitals (MO), and L-Click “OK” to observe the contour plots of the orbitals. 11.L-Click “Compute” and select “Vibrations”. 12.Make sure that “Rendering/Sphere” is on; L-Click “Compute” and select “Vibrational Spectrum”. Note that frequencies of different vibrational modes. 13.Turn on “Animate vibrations”, select one of the three modes, and L-Click “OK”. Water molecule begins to vibrate. To suspend the animation, L-Click on “Cancel”.
, and L-Click OK to observe the contour plots of the orbitals. 11.L-Click Compute and select Vibrations . 12.Make sure that Rendering/Sphere is on; L-Click Compute and select Vibrational Spectrum . Note that frequencies of different vibrational modes. 13.Turn on Animate vibrations , select one of the three modes, and L-Click OK . Water molecule begins to vibrate. To suspend the animation, L-Click on Cancel ..")
101
The Hartree-Fock treatment of H 2 + e-e- + e-e-

102
f 1 = 1 (1) 2 (2) f 2 = 1 (2) 2 (1) = c 1 f 1 + c 2 f 2 H 11 - WH 12 - S W H 21 - S WH 22 - W H 11 = H 22 = H 12 = H 21 = S = [ = S 2 ] The Heitler-London ground-state wave function {[ 1 (1) 2 (2) + 1 (2) 2 (1)]/ 2(1+S) 1/2 } [ (1) (2) (2) (1)]/ 2 = 0 The Valence-Bond Treatment of H 2
![f 1 = 1 (1) 2 (2) f 2 = 1 (2) 2 (1) = c 1 f 1 + c 2 f 2 H 11 - WH 12 - S W H 21 - S WH 22 - W H 11 = H 22 = H 12 = H 21 = S = [ = S 2 ] The Heitler-London ground-state wave function {[ 1 (1) 2 (2) + 1 (2) 2 (1)]/ 2(1+S) 1/2 } [ (1) (2) (2) (1)]/ 2 = 0 The Valence-Bond Treatment of H 2](http://images.slideplayer.com/16/5051037/slides/slide_102.jpg "f 1 = 1 (1) 2 (2) f 2 = 1 (2) 2 (1) = c 1 f 1 + c 2 f 2 H 11 - WH 12 - S W H 21 - S WH 22 - W H 11 = H 22 = H 12 = H 21 = S = [ = S 2 ] The Heitler-London ground-state wave function {[ 1 (1) 2 (2) + 1 (2) 2 (1)]/ 2(1+S) 1/2 } [ (1) (2) (2) (1)]/ 2 = 0 The Valence-Bond Treatment of H 2")
103
Comparison of the HF and VB Treatments HF LCAO-MO wave function for H 2 [ 1 (1) + 2 (1)] [ 1 (2) + 2 (2)] = 1 (1) 1 (2) + 1 (1) 2 (2) + 2 (1) 1 (2) + 2 (1) 2 (2) H H H H H H H H VB wave function for H 2 1 (1) 2 (2) + 2 (1) 1 (2) H H H H
![Comparison of the HF and VB Treatments HF LCAO-MO wave function for H 2 [ 1 (1) + 2 (1)] [ 1 (2) + 2 (2)] = 1 (1) 1 (2) + 1 (1) 2 (2) + 2 (1) 1 (2) + 2 (1) 2 (2) H H H H H H H H VB wave function for H 2 1 (1) 2 (2) + 2 (1) 1 (2) H H H H](http://images.slideplayer.com/16/5051037/slides/slide_103.jpg "Comparison of the HF and VB Treatments HF LCAO-MO wave function for H 2 [ 1 (1) + 2 (1)] [ 1 (2) + 2 (2)] = 1 (1) 1 (2) + 1 (1) 2 (2) + 2 (1) 1 (2) + 2 (1) 2 (2) H H H H H H H H VB wave function for H 2 1 (1) 2 (2) + 2 (1) 1 (2) H H H H")
104
At large distance, the system becomes H............ H MO: 50% H............ H 50% H +............ H VB: 100% H............ H The VB is computationally expensive and requires chemical intuition in implementation. The Generalized valence-bond (GVB) method is a variational method, and thus computationally feasible. (William A. Goddard III)
 method is a variational method, and thus computationally feasible. (William A. Goddard III).")
105
The Heitler-London ground-state wave function

106
Assignment 8.4, 8.10, 8.12b, 8.40, 10.5, 10.6, 10.7, 10.8, 11.37, 13.37

107
Electron Correlation Human Repulsive Correlation

108
Electron Correlation: avoiding each other Two reasons of the instantaneous correlation: (1) Pauli Exclusion Principle (HF includes the effect) (2) Coulomb repulsion (not included in the HF) Beyond the Hartree-Fock Configuration Interaction (CI)* Perturbation theory* Coupled Cluster Method Density functional theory
 Pauli Exclusion Principle (HF includes the effect) (2) Coulomb repulsion (not included in the HF) Beyond the Hartree-Fock Configuration Interaction (CI)* Perturbation theory* Coupled Cluster Method Density functional theory")
109
-e -e r 12 r 2 r 1 +2e H = - (h 2 /2m e ) 1 2 - 2e 2 /r 1 - (h 2 /2m e ) 2 2 - 2e 2 /r 2 + e 2 /r 12 H 1 0 H 2 0 H’
 e 2 /r 1 - (h 2 /2m e ) e 2 /r 2 + e 2 /r 12 H 1 0 H 2 0 H’")
110
H 0 = H 1 0 + H 2 0 (0) (1,2) = F 1 (1) F 2 (2) H 1 0 F 1 (1) = E 1 F 1 (1) H 2 0 F 2 (1) = E 2 F 2 (1) E 1 = -2e 2 /n 1 2 a 0 n 1 = 1, 2, 3,... E 2 = -2e 2 /n 2 2 a 0 n 2 = 1, 2, 3,... (0) (1,2) = (1/ 2 a 0 ) 3/2 exp(-2r 1 /a 0 ) (1/ 2 a 0 ) 3/2 exp(-2r 1 /a 0 ) E (0) = 4e 2 /a 0 E (1) = = 5e 2 /4a 0 E E (0) + E (1) = -108.8 + 34.0 = -74.8 (eV) [compared with exp. -79.0 eV] Ground state wave function
 (1,2) = (1/ 2 a 0 ) 3/2 exp(-2r 1 /a 0 ) (1/ 2 a 0 ) 3/2 exp(-2r 1 /a 0 ) E (0) = 4e 2 /a 0 E (1) = = 5e 2 /4a 0 E E (0) + E (1) = = (eV) [compared with exp eV] Ground state wave function.")
111
H = H 0 + H’ H 0 n (0) = E n (0) n (0) n (0) is an eigenstate for unperturbed system H’ is small compared with H 0 Nondegenerate Perturbation Theory (for Non-Degenerate Energy Levels)
 = E n (0) n (0) n (0) is an eigenstate for unperturbed system H’ is small compared with H 0 Nondegenerate Perturbation Theory (for Non-Degenerate Energy Levels)")
112
H( = H 0 + H’ H n = E n n n n n n k n (k) n n n n k n (k) the original Hamiltonian Introducing a parameter n n n n n (k) n n n n n (k) Where, = 0, j=1,2,...,k,...
 n n n n k n (k) the original Hamiltonian Introducing a parameter n n n n n (k) n n n n n (k) Where, = 0, j=1,2,...,k,...")
113
H n = E n n solving for E n n H n H’ n = E n n n n solving for E n n H n H’ n = E n n n n n n solving for E n n

114
Multiplied m (0) from the left and integrate, + = E n E n mn [E m E n + = E n mn For m = n, For m n, = / [E n E m If we expand n (1) = c nm m (0), c nm = / [E n E m for m n; c nn = 0. n (1) = m / [E n E m m (0) Eq.(2) The first order: E n Eq.(1)
 = m / [E n E m m (0) Eq.(2) The first order: E n Eq.(1).")
115
The second order: + = < m (0) n (2) >E n E n E n mn Set m = n, we have E n = m n | m (0) H' n (0) >| 2 / [E n E m q.(3)
 n (2) >E n E n E n mn Set m = n, we have E n = m n | m (0) H n (0) >| 2 / [E n E m q.(3)")
116
a. Eq.(2) shows that the effect of the perturbation on the wave function n (0) is to mix in contributions from the other zero-th order states m (0) m n. Because of the factor 1/(E n (0) -E m (0) ), the most important contributions to the n (1) come from the states nearest in energy to state n. b. To evaluate the first-order correction in energy, we need only to evaluate a single integral H’ nn ; to evaluate the second-order energy correction, we must evalute the matrix elements H’ between the n-th and all other states m. c. The summation in Eq.(2), (3) is over all the states, not the energy levels. Discussion: (Text Book: page 522-527)
 shows that the effect of the perturbation on the wave function n (0) is to mix in contributions from the other zero-th order states m (0) m n. Because of the factor 1/(E n (0) -E m (0) ), the most important contributions to the n (1) come from the states nearest in energy to state n. b. To evaluate the first-order correction in energy, we need only to evaluate a single integral H’ nn ; to evaluate the second-order energy correction, we must evalute the matrix elements H’ between the n-th and all other states m. c. The summation in Eq.(2), (3) is over all the states, not the energy levels. Discussion: (Text Book: page ).")
117
Moller-Plesset (MP) Perturbation Theory The MP unperturbed Hamiltonian H 0 H 0 = m F(m) where F(m) is the Fock operator for electron m. And thus, the perturbation H ’ H ’ = H - H 0 Therefore, the unperturbed wave function is simply the Hartree-Fock wave function . Ab initio methods: MP2, MP4

118
Example One: Consider the one-particle, one-dimensional system with potential-energy function V = b for L/4 < x < 3L/4, V = 0 for 0 < x L/4 & 3L/4 x < L and V = elsewhere. Assume that the magnitude of b is small, and can be treated as a perturbation. Find the first-order energy correction for the ground and first excited states. The unperturbed wave functions of the ground and first excited states are 1 = (2/L) 1/2 sin( x/L) and 2 = (2/L) 1/2 sin(2 x/L), respectively.
 1/2 sin( x/L) and 2 = (2/L) 1/2 sin(2 x/L), respectively..")
119
Example Two: As the first step of the Moller-Plesset perturbation theory, Hartree-Fock method gives the zeroth-order energy. Is the above statement correct? Example Three: Show that, for any perturbation H’, E 1 (0) + E 1 (1) E 1 where E 1 (0) and E 1 (1) are the zero-th order energy and the first order energy correction, and E 1 is the ground state energy of the full Hamiltonian H 0 + H’. Example Four: Calculate the bond orders of Li 2 and Li 2 +.
 + E 1 (1) E 1 where E 1 (0) and E 1 (1) are the zero-th order energy and the first order energy correction, and E 1 is the ground state energy of the full Hamiltonian H 0 + H’. Example Four: Calculate the bond orders of Li 2 and Li")
120
Perturbation Theory for a Degenerate Energy Level B / Hydrogen Atom n=3 3s, 3p x, 3p y, 3p z, 3d 1-5 n=2 2s, 2p x, 2p y, 2p z n=1 1s H = H 0 + H’ H 0 n (0) = E d (0) n (0) n=1,2,...,d H’ is small compared with H 0
 = E d (0) n (0) n=1,2,...,d H’ is small compared with H 0")
121
c nm = / [E n E m for 1 m, n d WRONG ! something very different ! (1)Apply the results of nondegenerate perturbation theory (2) What happened ? c 1 1 (0) + c 2 2 (0) +... + c d d (0) is an eigenstate for H 0 There are infinite number of such states that are degenerate.
Apply the results of nondegenerate perturbation theory (2) What happened . c 1 1 (0) + c 2 2 (0) c d d (0) is an eigenstate for H 0 There are infinite number of such states that are degenerate..")
122
When H’ is switched on, these states are no longer degenerate, and nondegenerate eigenstates of H 0 + H’ appear ! Therefore, even for zero-th order of eigenstates, there are sudden changes ! (3) Introducing a parameter H( = H 0 + H’ H n = E n n the original Hamiltonian n n n n k n (k) n d n n k n (k) n k c k k (0)
 Introducing a parameter H( = H 0 + H’ H n = E n n the original Hamiltonian n n n n k n (k) n d n n k n (k) n k c k k (0).")
123
H n H’ n = E d n n n solving for E n n n Multiplied m (0) from the left and integrate, + = E d E n [E m E d + = E n For 1 m d, n E m mn ] c n E m Assignment 2: 9.2, 9.4a, 9.9, 9.18, 9.24
![H n H’ n = E d n n n solving for E n n n Multiplied m (0) from the left and integrate, + = E d E n [E m E d + = E n For 1 m d, n E m mn ] c n E m Assignment 2: 9.2, 9.4a, 9.9, 9.18, 9.24](http://images.slideplayer.com/16/5051037/slides/slide_123.jpg "H n H’ n = E d n n n solving for E n n n Multiplied m (0) from the left and integrate, + = E d E n [E m E d + = E n For 1 m d, n E m mn ] c n E m Assignment 2: 9.2, 9.4a, 9.9, 9.18, 9.24")
124
Configuration Interaction (CI) + + …
 + + …")
125
Single Electron Excitation or Singly Excited

126
Double Electrons Excitation or Doubly Excited

127
Singly Excited Configuration Interaction (CIS): Changes only the excited states +
: Changes only the excited states +")
128
Doubly Excited CI (CID): Changes ground & excited states + Singly & Doubly Excited CI (CISD): Most Used CI Method
: Changes ground & excited states + Singly & Doubly Excited CI (CISD): Most Used CI Method")
129
Full CI (FCI): Changes ground & excited states + + +...
: Changes ground & excited states")
130
= e T (0) (0) : Hartree-Fock ground state wave function : Ground state wave function T = T 1 + T 2 + T 3 + T 4 + T 5 + … T n : n electron excitation operator Coupled-Cluster Method = T1T1
 (0) : Hartree-Fock ground state wave function : Ground state wave function T = T 1 + T 2 + T 3 + T 4 + T 5 + … T n : n electron excitation operator Coupled-Cluster Method = T1T1")
131
CCD = e T 2 (0) (0) : Hartree-Fock ground state wave function CCD : Ground state wave function T 2 : two electron excitation operator Coupled-Cluster Doubles (CCD) Method = T2T2
 (0) : Hartree-Fock ground state wave function CCD : Ground state wave function T 2 : two electron excitation operator Coupled-Cluster Doubles (CCD) Method = T2T2")
132
Complete Active Space SCF (CASSCF) Active space All possible configurations
 Active space All possible configurations")
133
Density-Functional Theory (DFT) Hohenberg-Kohn Theorem: Phys. Rev. 136, B864 (1964) The ground state electronic density (r) determines uniquely all possible properties of an electronic system (r) Properties P (e.g. conductance), i.e. P P[ (r)] Density-Functional Theory (DFT) E 0 = h 2 /2m e ) i dr e 2 (r) / r 1 dr 1 dr 2 e 2 /r 12 + E xc [ (r) ] Kohn-Sham Equation Ground State : Phys. Rev. 140, A1133 (1965) F KS i = i i F KS h 2 /2m e ) i i 2 e 2 / r 1 j J j + V xc V xc E xc [ (r) ] / (r) A popular exchange-correlation functional E xc [ (r) ] : B3LYP
 The ground state electronic density (r) determines uniquely all possible properties of an electronic system (r) Properties P (e.g. conductance), i.e. P P[ (r)] Density-Functional Theory (DFT) E 0 = h 2 /2m e ) i dr e 2 (r) / r 1 dr 1 dr 2 e 2 /r 12 + E xc [ (r) ] Kohn-Sham Equation Ground State : Phys. Rev. 140, A1133 (1965) F KS i = i i F KS h 2 /2m e ) i i 2 e 2 / r 1 j J j + V xc V xc E xc [ (r) ] / (r) A popular exchange-correlation functional E xc [ (r) ] : B3LYP.")
134
Time-Dependent Density-Functional Theory (TDDFT) Runge-Gross Extension: Phys. Rev. Lett. 52, 997 (1984) Time-dependent system (r,t) Properties P (e.g. absorption) TDDFT equation: exact for excited states Isolated system Open system Density-Functional Theory for Open System ??? Further Extension: X. Zheng, F. Wang & G.H. Chen (2005) Generalized TDDFT equation: exact for open systems
 Time-dependent system (r,t) Properties P (e.g. absorption) TDDFT equation: exact for excited states Isolated system Open system Density-Functional Theory for Open System . Further Extension: X. Zheng, F. Wang & G.H. Chen (2005) Generalized TDDFT equation: exact for open systems.")
135
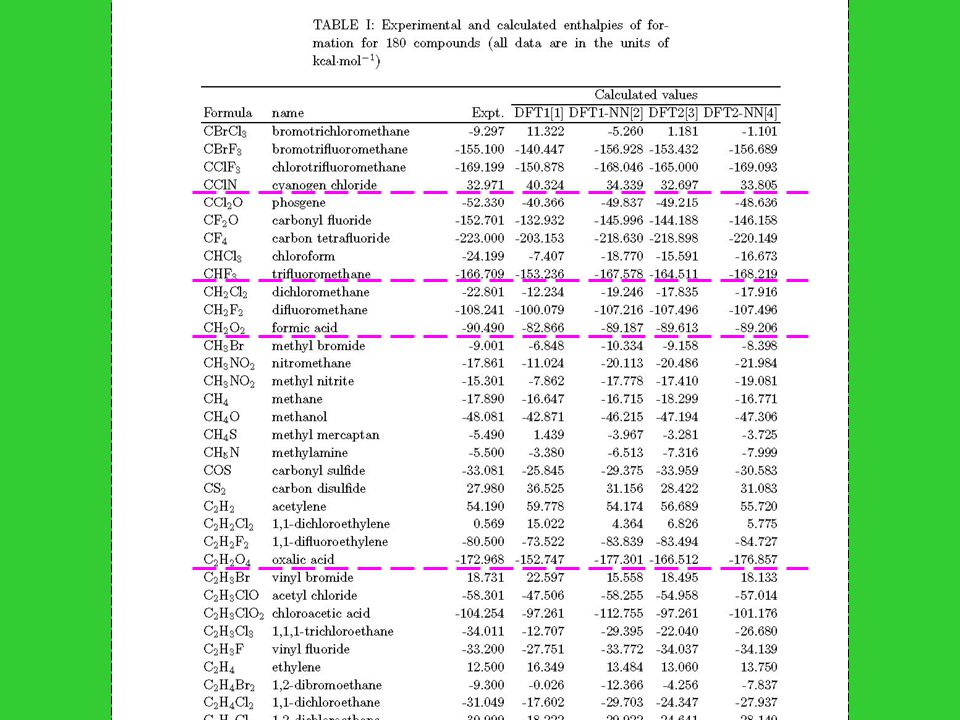
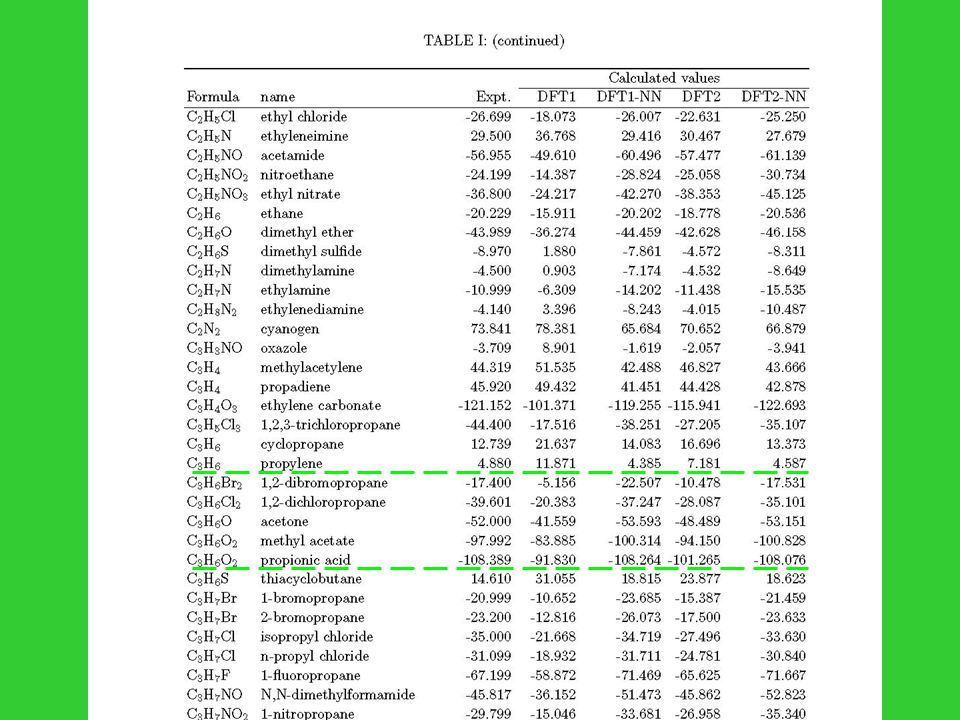
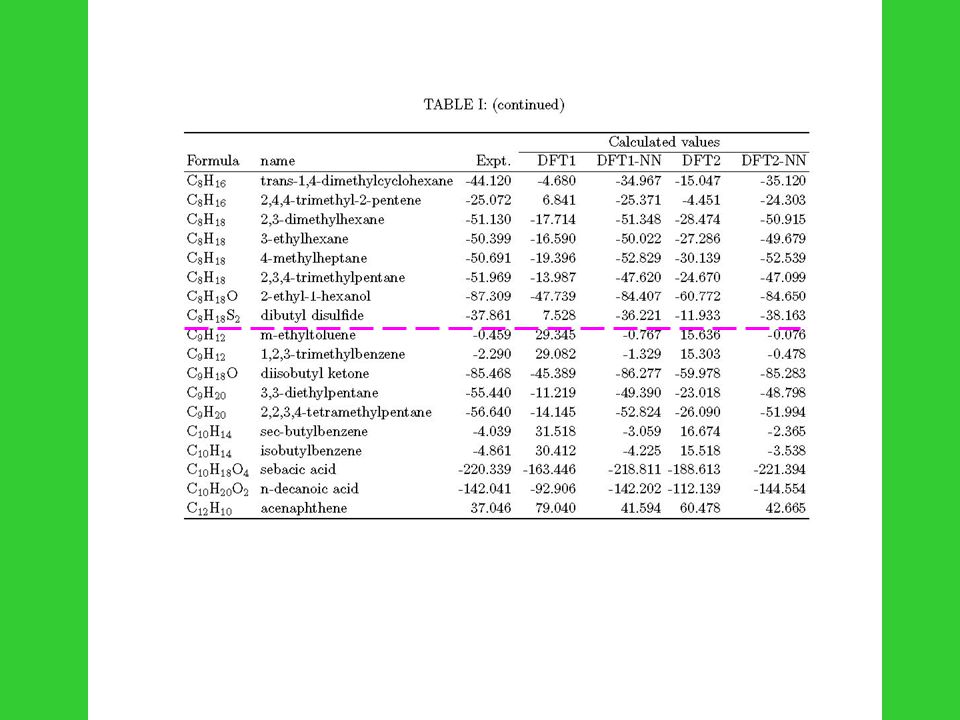
180 small- or medium-size organic molecules : 1. C.L. Yaws, Chemical Properties Handbook, (McGraw-Hill, New York, 1999) 2. D.R. Lide, CRC Handbook of Chemistry and Physics, 3 rd ed. (CRC Press, Boca Raton, FL, 2000) 3. J.B. Pedley, R.D. Naylor, S.P. Kirby, Thermochemical data of organic compunds, 2 nd ed. (Chapman and Hall, New York, 1986) Differences of heat of formation in three references for same compound are less than 1 kcal/mol; and error bars are all less than 1kcal/mol
 2. D.R. Lide, CRC Handbook of Chemistry and Physics, 3 rd ed. (CRC Press, Boca Raton, FL, 2000) 3. J.B. Pedley, R.D. Naylor, S.P. Kirby, Thermochemical data of organic compunds, 2 nd ed. (Chapman and Hall, New York, 1986) Differences of heat of formation in three references for same compound are less than 1 kcal/mol; and error bars are all less than 1kcal/mol.")
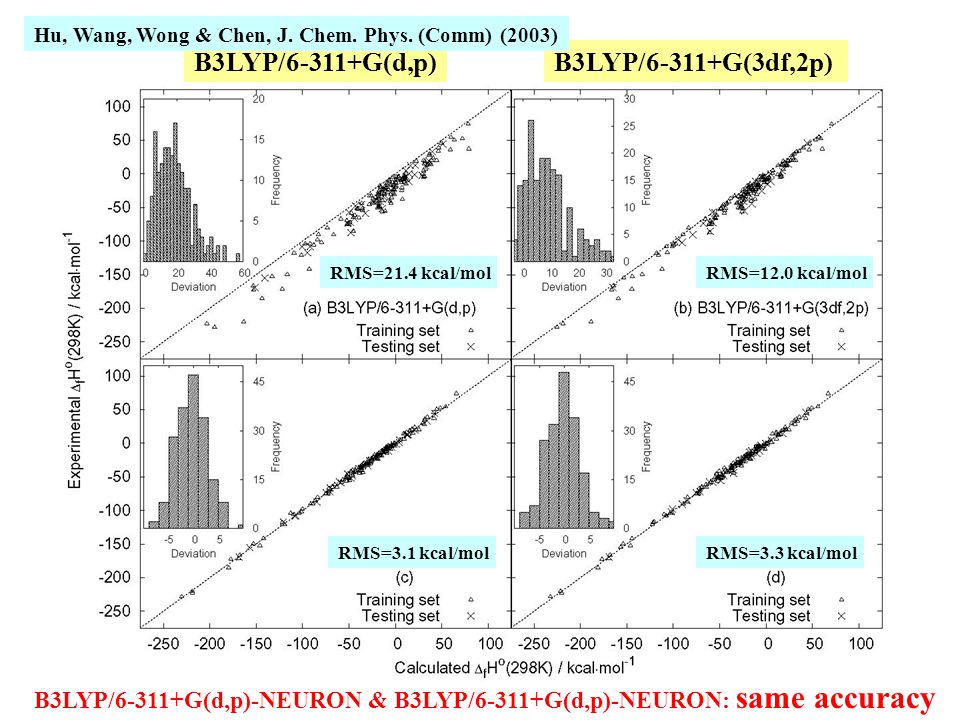
140
B3LYP/6-311+G(d,p)B3LYP/6-311+G(3df,2p) RMS=21.4 kcal/molRMS=12.0 kcal/mol RMS=3.1 kcal/molRMS=3.3 kcal/mol B3LYP/6-311+G(d,p)-NEURON & B3LYP/6-311+G(d,p)-NEURON: same accuracy Hu, Wang, Wong & Chen, J. Chem. Phys. (Comm) (2003)
 (2003).")
141
Ground State Excited State CPU Time Correlation Geometry Size Consistent (CHNH,6-31G*) HFSCF 1 0 OK DFT ~1 CIS <10 OK CISD 17 80-90% (20 electrons) CISDTQ very large 98-99% MP2 1.5 85-95% (DZ+P) MP4 5.8 >90% CCD large >90% CCSDT very large ~100%
 HFSCF 1 0 OK DFT ~1 CIS <10 OK CISD % (20 electrons) CISDTQ very large 98-99% MP2 % (DZ+P) MP4 5.8 >90% CCD large >90% CCSDT very large ~100% ")
142
Relativistic Effects Speed of 1s electron: Zc / 137 Heavy elements have large Z, thus relativistic effects are important. Dirac Equation: Relativistic Hartree-Fock w/ Dirac-Fock operator; or Relativistic Kohn-Sham calculation; or Relativistic effective core potential (ECP).
..")
143
(1) Neglect or incomplete treatment of electron correlation (2) Incompleteness of the Basis set (3) Relativistic effects (4) Deviation from the Born-Oppenheimer approximation Four Sources of error in ab initio Calculation
 Neglect or incomplete treatment of electron correlation (2) Incompleteness of the Basis set (3) Relativistic effects (4) Deviation from the Born-Oppenheimer approximation Four Sources of error in ab initio Calculation")
144
Semiempirical Molecular Orbital Calculation Extended Huckel MO Method (Wolfsberg, Helmholz, Hoffman) Independent electron approximation Schrodinger equation for electron i H val = i H eff (i) H eff (i) = -(h 2 /2m) i 2 + V eff (i) H eff (i) i = i i
 Independent electron approximation Schrodinger equation for electron i H val = i H eff (i) H eff (i) = -(h 2 /2m) i 2 + V eff (i) H eff (i) i = i i")
145
LCAO-MO: i = r c ri r s ( H eff rs - i S rs ) c si = 0 H eff rs r H eff s S rs r s Parametrization: H eff rr r H eff r minus the valence-state ionization potential (VISP)
 c si = 0 H eff rs r H eff s S rs r s Parametrization: H eff rr r H eff r minus the valence-state ionization potential (VISP)")
146
Atomic Orbital Energy VISP ---------------e 5 -e 5 ---------------e 4 -e 4 ---------------e 3 -e 3 ---------------e 2 -e 2 ---------------e 1 -e 1 H eff rs = ½ K (H eff rr + H eff ss ) S rs K:1 3
 S rs K:1 3")
147
CNDO, INDO, NDDO (Pople and co-workers) Hamiltonian with effective potentials H val = i [ -(h 2 /2m) i 2 + V eff (i) ] + i j>i e 2 / r ij two-electron integral: (rs|tu) = CNDO: complete neglect of differential overlap (rs|tu) = rs tu (rr|tt) rs tu rt
![CNDO, INDO, NDDO (Pople and co-workers) Hamiltonian with effective potentials H val = i [ -(h 2 /2m) i 2 + V eff (i) ] + i j>i e 2 / r ij two-electron integral: (rs|tu) = CNDO: complete neglect of differential overlap (rs|tu) = rs tu (rr|tt) rs tu rt](http://images.slideplayer.com/16/5051037/slides/slide_147.jpg "CNDO, INDO, NDDO (Pople and co-workers) Hamiltonian with effective potentials H val = i [ -(h 2 /2m) i 2 + V eff (i) ] + i j>i e 2 / r ij two-electron integral: (rs|tu) = CNDO: complete neglect of differential overlap (rs|tu) = rs tu (rr|tt) rs tu rt")
148
INDO: intermediate neglect of differential overlap (rs|tu) = rs tu (rr|tt) when r, s, t & u not on same atom; (rs|tu) 0 when r, s, t and u are on the same atom. NDDO: neglect of diatomic differential overlap (rs|tu) = 0 if r and s (or t and u) are not on the same atom. CNDO, INDO are parametrized so that the overall results fit well with the results of minimal basis ab initio Hartree-Fock calculation. CNDO/S, INDO/S are parametrized to predict optical spectra.
 = 0 if r and s (or t and u) are not on the same atom. CNDO, INDO are parametrized so that the overall results fit well with the results of minimal basis ab initio Hartree-Fock calculation. CNDO/S, INDO/S are parametrized to predict optical spectra..")
149
PRDDO H = i [ -(h 2 /2m) i 2 + V eff (i) ] + i j>i e 2 / r ij Basis set: the minimum basis set (STO-3G) PRDDO: partial retention of diatomic differential overlap (rs|tu) = 0 if r and s (and t and u) are different basis functions.
![PRDDO H = i [ -(h 2 /2m) i 2 + V eff (i) ] + i j>i e 2 / r ij Basis set: the minimum basis set (STO-3G) PRDDO: partial retention of diatomic differential overlap (rs|tu) = 0 if r and s (and t and u) are different basis functions.](http://images.slideplayer.com/16/5051037/slides/slide_149.jpg "PRDDO H = i [ -(h 2 /2m) i 2 + V eff (i) ] + i j>i e 2 / r ij Basis set: the minimum basis set (STO-3G) PRDDO: partial retention of diatomic differential overlap (rs|tu) = 0 if r and s (and t and u) are different basis functions.")
150
MINDO, MNDO, AM1, PM3 (Dewar and co-workers, University of Texas, Austin) MINDO: modified INDO MNDO: modified neglect of diatomic overlap AM1: Austin Model 1 PM3: MNDO parametric method 3 MINDO, MNDO, AM1 & PM3: *based on INDO & NDDO *reproduce the binding energy
 MINDO: modified INDO MNDO: modified neglect of diatomic overlap AM1: Austin Model 1 PM3: MNDO parametric method 3 MINDO, MNDO, AM1 & PM3: *based on INDO & NDDO *reproduce the binding energy")
151
Key: How to approximate ? Fock Matrix MNDO-PM3 (using NDDO) Semiempirical M.O. Method
 Semiempirical M.O. Method")
152
Where, : the ionization potential One centre integrals: (given) Core-electron attraction: (given) :characteristic of monopole, dipole, quadrupole :charge separations
 Core-electron attraction: (given) :characteristic of monopole, dipole, quadrupole :charge separations")
153
Molecular Mechanics (MM) Method F = Ma F : Force Field
 Method F = Ma F : Force Field")
154
Molecular Mechanics Force Field Bond Stretching Term Bond Angle Term Torsional Term Non-Bonding Terms: Electrostatic Interaction & van der Waals Interaction C 2 H 3 Cl

155
Bond Stretching Potential E b = 1/2 k b ( l) 2 where, k b : stretch force constant l : difference between equilibrium & actual bond length Two-body interaction
 2 where, k b : stretch force constant l : difference between equilibrium & actual bond length Two-body interaction")
156
Bond Angle Deformation Potential E a = 1/2 k a ( ) 2 where, k a : angle force constant : difference between equilibrium & actual bond angle Three-body interaction
 2 where, k a : angle force constant : difference between equilibrium & actual bond angle Three-body interaction")
157
Periodic Torsional Barrier Potential E t = (V/2) (1+ cosn ) where, V : rotational barrier : torsion angle n : rotational degeneracy Four-body interaction
 (1+ cosn ) where, V : rotational barrier : torsion angle n : rotational degeneracy Four-body interaction")
158
Non-bonding interaction van der Waals interaction for pairs of non-bonded atoms Coulomb potential for all pairs of charged atoms

159
MM Force Field Types MM2Small molecules AMBERPolymers CHAMMPolymers BIOPolymers OPLSSolvent Effects

160
CHAMM FORCE FIELD FILE

162
/Ao/Ao /(kcal/mol)
")
163
/(kcal/mol/A o2 ) /Ao/Ao
 /Ao/Ao")
164
/(kcal/mol/rad 2 ) /deg
 /deg")
165
/(kcal/mol)/deg
/deg")
166
Algorithms for Molecular Dynamics x(t+ t) = x(t) + (dx/dt) t Fourth-order Runge-Kutta method: x(t+ t) = x(t) + (1/6) (s 1 +2s 2 +2s 3 +s 4 ) t +O( t 5 ) s 1 = dx/dt s 2 = dx/dt [w/ t=t+ t/2, x = x(t)+s 1 t/2] s 3 = dx/dt [w/ t=t+ t/2, x = x(t)+s 2 t/2] s 4 = dx/dt [w/ t=t+ t, x = x(t)+s 3 t] Very accurate but slow!
![Algorithms for Molecular Dynamics x(t+ t) = x(t) + (dx/dt) t Fourth-order Runge-Kutta method: x(t+ t) = x(t) + (1/6) (s 1 +2s 2 +2s 3 +s 4 ) t +O( t 5 ) s 1 = dx/dt s 2 = dx/dt [w/ t=t+ t/2, x = x(t)+s 1 t/2] s 3 = dx/dt [w/ t=t+ t/2, x = x(t)+s 2 t/2] s 4 = dx/dt [w/ t=t+ t, x = x(t)+s 3 t] Very accurate but slow!](http://images.slideplayer.com/16/5051037/slides/slide_166.jpg "Algorithms for Molecular Dynamics x(t+ t) = x(t) + (dx/dt) t Fourth-order Runge-Kutta method: x(t+ t) = x(t) + (1/6) (s 1 +2s 2 +2s 3 +s 4 ) t +O( t 5 ) s 1 = dx/dt s 2 = dx/dt [w/ t=t+ t/2, x = x(t)+s 1 t/2] s 3 = dx/dt [w/ t=t+ t/2, x = x(t)+s 2 t/2] s 4 = dx/dt [w/ t=t+ t, x = x(t)+s 3 t] Very accurate but slow!")
167
Algorithms for Molecular Dynamics Verlet Algorithm: x(t+ t) = x(t) + (dx/dt) t + (1/2) d 2 x/dt 2 t 2 +... x(t - t) = x(t) - (dx/dt) t + (1/2) d 2 x/dt 2 t 2 -... x(t+ t) = 2x(t) - x(t - t) + d 2 x/dt 2 t 2 + O( t 4 ) Efficient & Commonly Used!
 = x(t) - (dx/dt) t + (1/2) d 2 x/dt 2 t x(t+ t) = 2x(t) - x(t - t) + d 2 x/dt 2 t 2 + O( t 4 ) Efficient & Commonly Used!.")
168
Calculated Properties Structure, Geometry Energy & Stability Vibration Frequency & Mode Real Time Dynamics

169
Summary Hamiltonian H = ( h 2 /2m h 2 /2m e ) i i 2 + Z Z e r i e 2 /r i i j e 2 /r ij Consider a system whose Hamiltonian operator H is time independent and whose lowest-energy eigenvalue is E 1. If is any well-behaved function that satisfies the boundary conditions of the problem, then * H d * d E 1 The variation theorem (1) Construct a wave function (c 1,c 2, ,c m ) (2) Calculate the energy of : E E (c 1,c 2, ,c m ) (3) Choose {c j * } (i=1,2, ,m) so that E is minimum Variational Method
 Construct a wave function (c 1,c 2, ,c m ) (2) Calculate the energy of : E E (c 1,c 2, ,c m ) (3) Choose {c j * } (i=1,2, ,m) so that E is minimum Variational Method.")
170
Extension of Variation Method For a wave function which is orthogonal to the ground state wave function 1, i.e. d * 1 = 0 E = d * H / d * > E 2 the first excited state energy The Pauli principle two electrons cannot be in the same state the wave function of a system of electrons must be antisymmetric with respect to interchanging of any two electrons. Slater determinant f H 2 : ! [ !

171
f(1)+ J 2 (1) K 2 (1) 1 (1) 1 1 (1) f(2)+ J 1 (2) K 1 (2) 2 (2) 2 2 (2) F(1) f(1)+ J 2 (1) K 2 (1) Fock operator for 1 F(2) f(2)+ J 1 (2) K 1 (2) Fock operator for 2 Hartree-Fock Equation: Fock Operator: LCAO-MO: c 1 1 + c 2 2 Molecule Bond order De/eV H 2 + 1/2 2.79 H 2 1 4.75 He 2 + 1/2 1.08 He 2 0 0.0009 Li 2 1 1.07 Be 2 0 0.10 C 2 2 6.3 N 2 + 1/2 8.85 N 2 3 9.91 O 2 2 5.21 Express Hartree-Fock energy in terms of f i, J ij & K ij
+ J 2 (1) K 2 (1) 1 (1) 1 1 (1) f(2)+ J 1 (2) K 1 (2) 2 (2) 2 2 (2) F(1) f(1)+ J 2 (1) K 2 (1) Fock operator for 1 F(2) f(2)+ J 1 (2) K 1 (2) Fock operator for 2 Hartree-Fock Equation: Fock Operator: LCAO-MO: c 1 1 + c 2 2 Molecule Bond order De/eV H 2 + 1/ H He 2 + 1/ He Li Be C N 2 + 1/ N O Express Hartree-Fock energy in terms of f i, J ij & K ij")
172
Basis set of GTFs STO-3G, 3-21G, 4-31G, 6-31G, 6-31G*, 6-31G** ------------------------------------------------------------------------------------- complexity & accuracy # HF/6-31G(d) Route section water energy Title 0 1 Molecule Specification O -0.464 0.177 0.0 (in Cartesian coordinates H -0.464 1.137 0.0 H 0.441 -0.143 0.0 Gaussian 98 Input file Comparison of the HF and VB Treatments Electron Correlation
 Route section water energy Title 0 1 Molecule Specification O (in Cartesian coordinates H H Gaussian 98 Input file Comparison of the HF and VB Treatments Electron Correlation")
173
Beyond the Hartree-Fock Configuration Interaction (CI)* Perturbation theory* Coupled Cluster Method Density functional theory E n Moller-Plesset (MP) Perturbation Theory The MP unperturbed Hamiltonian H 0 H 0 = m F(m) where F(m) is the Fock operator for electron m. And thus, the perturbation H ’ H ’ = H - H 0

174
Ground State Excited State CPU Time Correlation Geometry Size Consistent (CH 3 NH 2,6-31G*) HFSCF 1 0 OK DFT ~1 CIS <10 OK CISD 17 80-90% (20 electrons) CISDTQ very large 98-99% MP2 1.5 85-95% (DZ+P) MP4 5.8 >90% CCD large >90% CCSDT very large ~100%
 HFSCF 1 0 OK DFT ~1 CIS <10 OK CISD % (20 electrons) CISDTQ very large 98-99% MP2 % (DZ+P) MP4 5.8 >90% CCD large >90% CCSDT very large ~100% ")
Similar presentations


















! Hψ = Eψ A differential (operator) eigenvalue equation H.>")





