Download presentation
Presentation is loading. Please wait.

1
Biochemical Tool Electrophoresis Hybridization

2
1.Molecules are separated by electric force 2.F = qE : where q is net charge, E is electric field strength 3.The velocity is encountered by friction 4.qE = fv : where f is frictional force, v is velocity 5.Therefore, mobility per unit field (U) = v/q = q/f = q/6p r : where is viscosity of supporting medium, r is radius of sphere molecule + - + - - - -+ E F f v q Electrophoresis Electro = flow of electricity, Phoresis= to carry across (from the Greek)
 = v/q = q/f = q/6p r : where is viscosity of supporting medium, r is radius of sphere molecule E F f v q Electrophoresis Electro = flow of electricity, Phoresis= to carry across (from the Greek)")
3
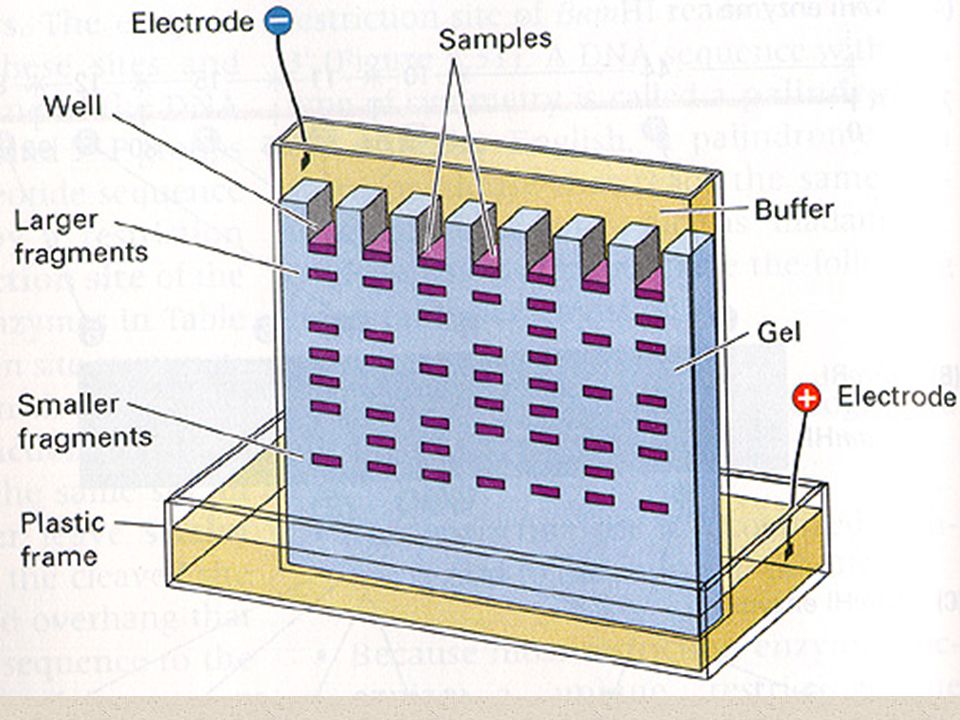
Definition The separation of charged molecules using their different rates of migration in an electrical field + Samples Separating Gel - FACTORS INFLUENCING SEPARATION Charge Density on Molecules - Difference between pH Molecular Size and Shape

4
Factors affected the mobility of molecules 1. Molecular factors Charge Size Shape 2. Environment factors Electric field strength Supporting media (pore: sieving effect) Running buffer - + Electrophoresis
 Running buffer - + Electrophoresis.")
6
Types of supporting media Paper Agarose gel (Agarose gel electrophoresis) Polyacrylamide gel (PAGE) pH gradient (Isoelectric focusing electrophoresis) Cellulose acetate
 Polyacrylamide gel (PAGE) pH gradient (Isoelectric focusing electrophoresis) Cellulose acetate")
7
Gel electrophoresis A gel is a colloid, a suspension of tiny particles in a medium, occurring in a solid form, like gelatin Gel electrophoresis refers to the separation of charged particles located in a gel when an electric current is applied Charged particles can include DNA, amino acids, peptides

8
Poliakrialimida Polimer dari akrilamid Pori-porinya lebih kecil dari polimer agarosa Menghasilkan tingkat resolusi yang lebih tinggi Gel dibuat dengan menggunakan 2 lembaran kaca atau plastik mika

9
Poliakrialimida Penyangga: TBE Kegunaan: 1.Memisahkan DNA berukuran kecil (AFLP, SNP) 2.mengurutkan DNA 3.Memisahkan protein (perlu ditambah SDS, sehingga disebut SDS-PAGE: SDS poly acrylamide Gel Electrophoresis)
 2.mengurutkan DNA 3.Memisahkan protein (perlu ditambah SDS, sehingga disebut SDS-PAGE: SDS poly acrylamide Gel Electrophoresis)")
10
Pembuatan gel poliakrialimida Akrilamida + metilen bis akrilamida Ukuran pori ditentukan dengan menentukan konsentrasi akrilamida dan metilen bis akrilamidanya

11
Polyacrylamide Gels Acrylamide polymer; very stable gel can be made at a wide variety of concentrations gradient of concentrations: large variety of pore sizes (powerful sieving effect) Electrophoresis
 Electrophoresis")
12
Sodium Dodecyl Sulfate = Sodium Lauryl Sulfate: CH 3 (CH 2 ) 11 SO 3 - Na + Amphipathic molecule Strong detergent to denature proteins Binding ratio: 1.4 gm SDS/gm protein Charge and shape normalization SDS-Polyacrylamide Gel Electrophoresis (SDS-PAGE)
 11 SO 3 - Na + Amphipathic molecule Strong detergent to denature proteins Binding ratio: 1.4 gm SDS/gm protein Charge and shape normalization SDS-Polyacrylamide Gel Electrophoresis (SDS-PAGE)")
13
Electrophoresis Isoelectric Focusing Electrophoresis (IFE) -Separate molecules according to their isoelectric point (pI) -At isoelectric point (pI) molecule has no charge (q=0), hence molecule ceases -pH gradient medium
 -Separate molecules according to their isoelectric point (pI) -At isoelectric point (pI) molecule has no charge (q=0), hence molecule ceases -pH gradient medium")
14
Electrophoresis 2-dimensional Gel Electrophoresis -First dimension is IFE (separated by pI) -Second dimension is SDS-PAGE (separated by size) -So called 2D-PAGE -High throughput electrophoresis, high resolution -Core methods for “Proteomics”
 -Second dimension is SDS-PAGE (separated by size) -So called 2D-PAGE -High throughput electrophoresis, high resolution -Core methods for Proteomics")
15
2-dimensional Gel Electrophoresis Spot coordination -pI -MW

16
2-dimensional Gel Electrophoresis Application

17
Hybridization and Blotting

18
Hybridization

19
Can be DNA:DNA, DNA:RNA, or RNA:RNA (RNA is easily degraded) Dependent on the extent of complementation Dependent on temperature, salt concentration, and solvents Small changes in the above factors can be used to discriminate between different sequences (e.g., small mutations can be detected) Probes can be labeled with radioactivity, fluorescent dyes, enzymes, etc. Probes can be isolated or synthesized sequences

20
Oligonucleotide probes Single stranded DNA (usually 15-40 bp) Degenerate oligonucleotide probes can be used to identify genes encoding characterized proteins –Use amino acid sequence to predict possible DNA sequences –Hybridize with a combination of probes –TT(T/C) - TGG - ATG - GA(T/C) - TG(T/C) - could be used for FWMDC amino acid sequence Can specifically detect single nucleotide changes
 Degenerate oligonucleotide probes can be used to identify genes encoding characterized proteins –Use amino acid sequence to predict possible DNA sequences –Hybridize with a combination of probes –TT(T/C) - TGG - ATG - GA(T/C) - TG(T/C) - could be used for FWMDC amino acid sequence Can specifically detect single nucleotide changes")
21
Detection of Probes Probes can be labeled with radioactivity, fluorescent dyes, enzymes. Radioactivity is often detected by X-ray film (autoradiography) Fluorescent dyes can be detected by fluorometers, scanners Enzymatic activities are often detected by the production of dyes or light (x-ray film)
 Fluorescent dyes can be detected by fluorometers, scanners Enzymatic activities are often detected by the production of dyes or light (x-ray film).")
22
RNA Blotting (Northerns) RNA is separated by size on a denaturing agarose gel and then transferred onto a membrane (blot) Probe is hybridized to complementary sequences on the blot and excess probe is washed away Location of probe is determined by detection method (e.g., film, fluorometer )
 RNA is separated by size on a denaturing agarose gel and then transferred onto a membrane (blot) Probe is hybridized to complementary sequences on the blot and excess probe is washed away Location of probe is determined by detection method (e.g., film, fluorometer )")
23
Applications of RNA Blots Detect the expression level and transcript size of a specific gene in a specific tissue or at a specific time. Sometimes mutations do not affect coding regions but transcriptional regulatory sequences (e.g., UAS/URS, promoter, splice sites, copy number, transcript stability, etc.)
.")
24
Western Blot Highly specific qualitative test Can determine if above or below threshold Typically used for research Use denaturing SDS-PAGE –Solubilizes, removes aggregates & adventitious proteins are eliminated Components of the gel are then transferred to a solid support or transfer membrane Paper towel Transfer membrane Wet filter paper Paper towel weight

25
Western Blot Add monoclonal antibodies Rinse again Antibodies will bind to specified protein Stain the bound antibody for colour development It should look like the gel you started with if a positive reaction occurred Block membrane e.g. dried nonfat milk Rinse with ddH 2 O Add antibody against yours with a marker (becomes the antigen)
.")
26
Polymerase Chain Reaction (PCR)
")
27
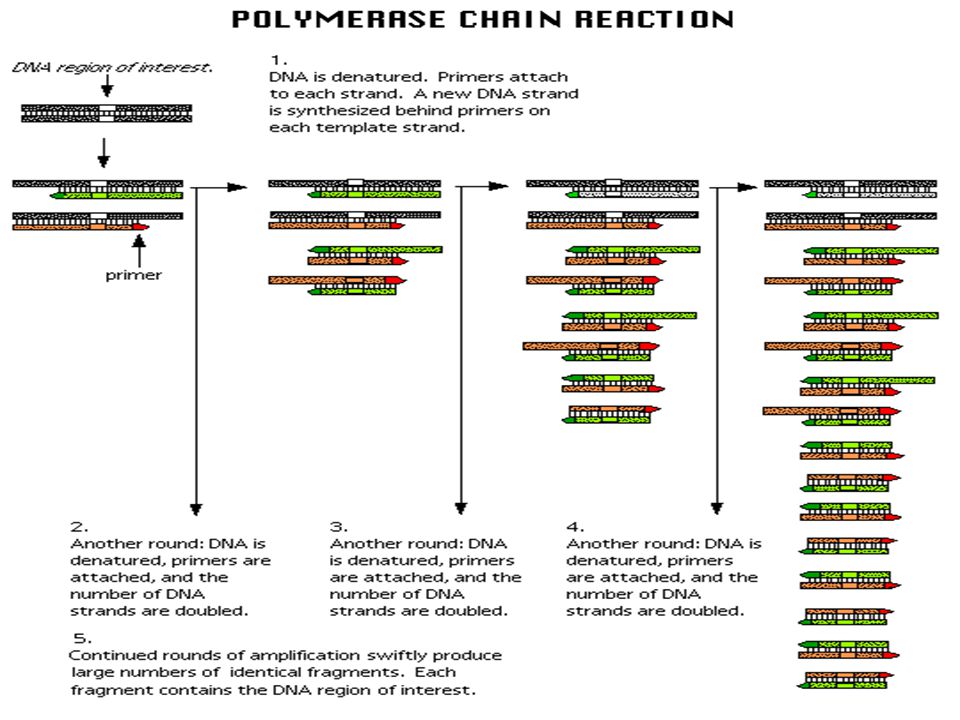
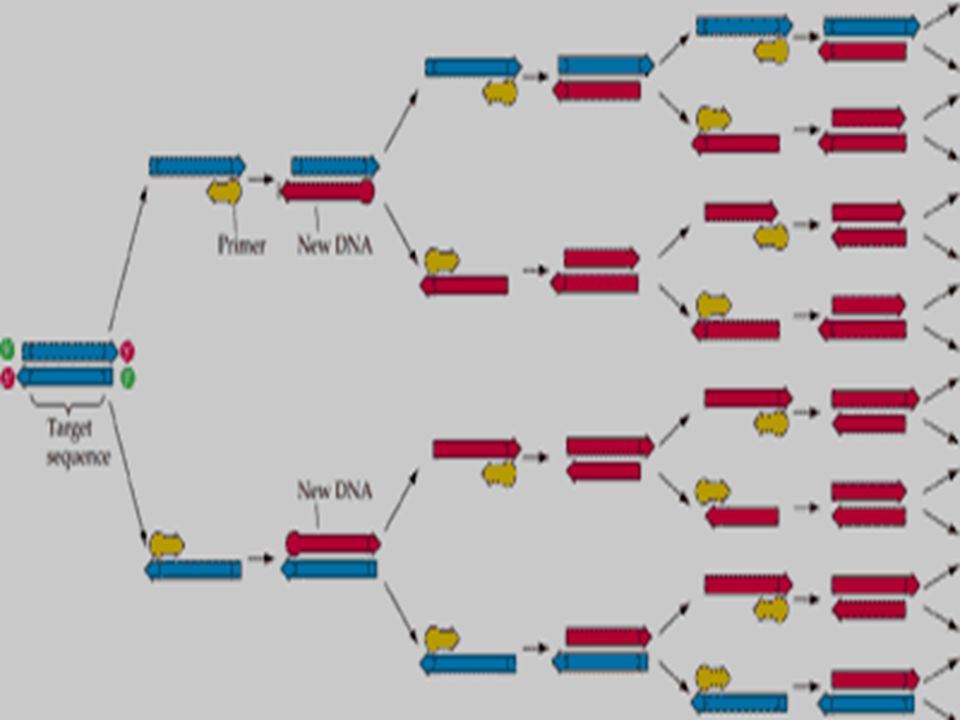
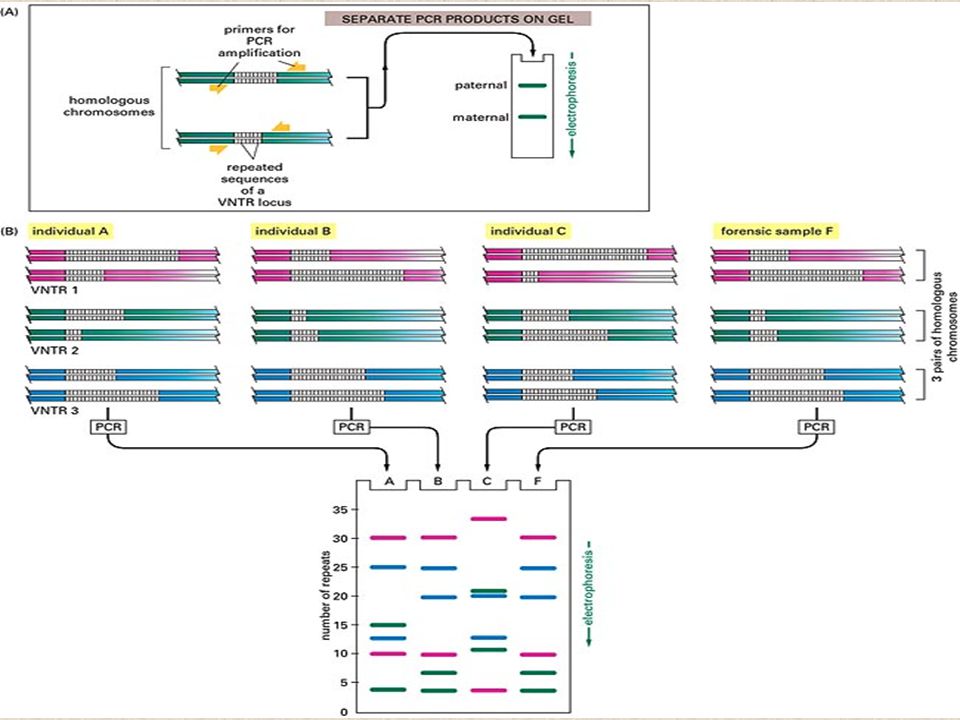
A simple rapid, sensitive and versatile in vitro method for selectively amplifying defined sequences/regions of DNA/RNA from an initial complex source of nucleic acid - generates sufficient for subsequent analysis and/or manipulation Amplification of a small amount of DNA using specific DNA primers (a common method of creating copies of specific fragments of DNA) DNA fragments are synthesized in vitro by repeated reactions of DNA synthesis (It rapidly amplifies a single DNA molecule into many billions of molecules) In one application of the technology, small samples of DNA, such as those found in a strand of hair at a crime scene, can produce sufficient copies to carry out forensic tests. Each cycle the amount of DNA doubles PCR

28
Ability to generate identical high copy number DNAs made possible in the 1970s by recombinant DNA technology (i.e., cloning). Cloning DNA is time consuming and expensive Probing libraries can be like hunting for a needle in a haystack. Requires only simple, inexpensive ingredients and a couple hours. Background on PCR

29
PCR, “discovered” in 1983 by Kary Mullis DNA template Primers (anneal to flanking sequences) DNA polymerase dNTPs Mg 2+ Buffer Can be performed by hand or in a machine called a thermal cycler. 1993: Nobel Prize for Chemistry Background on PCR

30
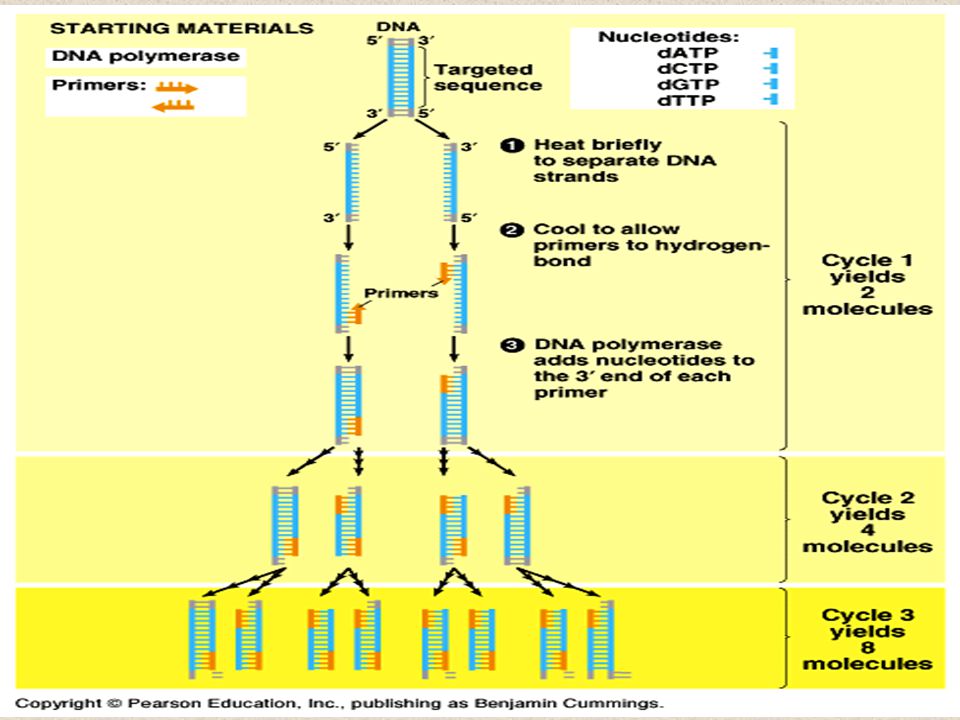
Three Steps Separation: Double Stranded DNA is denatured by heat into single strands. Short Primers for DNA replication are added to the mixture. DNA polymerase catalyzes the production of complementary new strands. Copying: the process is repeated for each new strand created All three steps are carried out in the same vial but at different temperatures

31
Step 1: Separation Combine Target Sequence, DNA primers template, dNTPs, Taq Polymerase Target Sequence: Usually fewer than 3000 bp –Identified by a specific pair of DNA primers- usually oligonucleotides that are about 20 nucleotides Heat to 95°C to separate strands (for 0.5-2 minutes) –Longer times increase denaturation but decrease enzyme and template
 –Longer times increase denaturation but decrease enzyme and template")
32
Magnesium as a Cofactor Stabilizes the reaction between: –oligonucleotides and template DNA –DNA Polymerase and template DNA

33
Heat: Denatures DNA by uncoiling the Double Helix strands.

34
Step 2: Priming Decrease temperature by 15-25 ° Primers anneal to the end of the strand 0.5-2 minutes Shorter time increases specificity but decreases yield Requires knowledge of the base sequences of the 3’ - end

35
Selecting a Primer Primer length Melting Temperature (T m ) Specificity Complementary Primer Sequences G/C content and Polypyrimidine (T, C) or polypurine (A, G) stretches 3’-end Sequence Single-stranded DNA
 Specificity Complementary Primer Sequences G/C content and Polypyrimidine (T, C) or polypurine (A, G) stretches 3’-end Sequence Single-stranded DNA")
36
Step 3: Polymerization Since the Taq polymerase works best at around 75 ° C (the temperature of the hot springs where the bacterium was discovered), the temperature of the vial is raised to 72-75 °C The DNA polymerase recognizes the primer and makes a complementary copy of the template which is now single stranded. Approximately 150 nucleotides/sec

37
Potential Problems with Taq Lack of proof-reading of newly synthesized DNA. Potentially can include di-Nucleotriphosphates (dNTPs) that are not complementary to the original strand. Errors in coding result Recently discovered thermostable DNA polymerases, Tth and Pfu, are less efficient, yet highly accurate.
 that are not complementary to the original strand. Errors in coding result Recently discovered thermostable DNA polymerases, Tth and Pfu, are less efficient, yet highly accurate..")
38
How PCR works: 1.Begins with DNA containing a sequence to be amplified and a pair of synthetic oligonucleotide primers that flank the sequence. 2.Next, denature the DNA at 94˚C. 3.Rapidly cool the DNA (37-65˚C) and anneal primers to complementary s.s. sequences flanking the target DNA. 4.Extend primers at 70-75˚C using a heat-resistant DNA polymerase (e.g., Taq polymerase derived from Thermus aquaticus). 5.Repeat the cycle of denaturing, annealing, and extension 20- 45 times to produce 1 million (2 20 ) to 35 trillion copies (2 45 ) of the target DNA. 6.Extend the primers at 70-75˚C once more to allow incomplete extension products in the reaction mixture to extend completely. 7. Cool to 4˚C and store or use amplified PCR product for analysis.
 and anneal primers to complementary s.s. sequences flanking the target DNA. 4.Extend primers at 70-75˚C using a heat-resistant DNA polymerase (e.g., Taq polymerase derived from Thermus aquaticus). 5.Repeat the cycle of denaturing, annealing, and extension times to produce 1 million (2 20 ) to 35 trillion copies (2 45 ) of the target DNA. 6.Extend the primers at 70-75˚C once more to allow incomplete extension products in the reaction mixture to extend completely. 7. Cool to 4˚C and store or use amplified PCR product for analysis..")
39
Example thermal cycler protocol used in lab: Step 1 7 min at 94˚CInitial Denature Step 2 45 cycles of: 20 sec at 94˚CDenature 20 sec at 64˚CAnneal 1 min at 72˚CExtension Step 3 7 min at 72˚CFinal Extension Step 4 Infinite hold at 4˚CStorage

40
The Polymerase Chain Reaction

44
PCR amplification Each cycle the oligo-nucleotide primers bind most all templates due to the high primer concentration The generation of mg quantities of DNA can be achieved in ~30 cycles (~ 4 hrs)
")
45
Starting nucleic acid - DNA/RNA Tissue, cells, blood, hair root, saliva, semen Thermo-stable DNA polymerase e.g. Taq polymerase Oligonucleotides Design them well! Buffer Tris-HCl (pH 7.6-8.0) Mg 2+ dNTPs (dATP, dCTP, dGTP, dTTP ) OPTIMISING PCR – THE REACTION COMPONENTS
 Mg 2+ dNTPs (dATP, dCTP, dGTP, dTTP ) OPTIMISING PCR – THE REACTION COMPONENTS.")
46
Tissue, cells, blood, hair root, saliva, semen Obtain the best starting material you can. Some can contain inhibitors of PCR, so they must be removed e.g. Haem in blood Good quality genomic DNA if possible Blood – consider commercially available reagents Qiagen– expense? Empirically determine the amount to add RAW MATERIAL

47
Number of options available Taq polymerase Pfu polymerase Tth polymerase How big is the product? 100bp 40-50kb What is end purpose of PCR? 1. Sequencing - mutation detection -. Need high fidelity polymerase -. integral 3’ 5' proofreading exonuclease activity 2. Cloning POLYMERASE

48
Length ~ 18-30 nucleotides (21 nucleotides) Base composition; 50 - 60% GC rich pairs should have equivalent Tms Tm = [(number of A+T residues) x 2 °C] + [(number of G+C residues) x 4 °C] Initial use Tm–5°C Avoid internal hairpin structures no secondary structure Avoid a T at the 3’ end Avoid overlapping 3’ ends – will form primer dimers Can modify 5’ ends to add restriction sites PRIMER DESIGN
![Length ~ nucleotides (21 nucleotides) Base composition; % GC rich pairs should have equivalent Tms Tm = [(number of A+T residues) x 2 °C] + [(number of G+C residues) x 4 °C] Initial use Tm–5°C Avoid internal hairpin structures no secondary structure Avoid a T at the 3’ end Avoid overlapping 3’ ends – will form primer dimers Can modify 5’ ends to add restriction sites PRIMER DESIGN](http://images.slideplayer.com/16/5048410/slides/slide_48.jpg "Length ~ nucleotides (21 nucleotides) Base composition; % GC rich pairs should have equivalent Tms Tm = [(number of A+T residues) x 2 °C] + [(number of G+C residues) x 4 °C] Initial use Tm–5°C Avoid internal hairpin structures no secondary structure Avoid a T at the 3’ end Avoid overlapping 3’ ends – will form primer dimers Can modify 5’ ends to add restriction sites PRIMER DESIGN")
49
Use specific programs OLIGO Medprobe PRIMER DESIGNER Sci. Ed software Also available on the internet http://www.hgmp.mrc.ac.uk/GenomeWeb/nuc-primer.html

50
Mg 2+ CONCENTRATION 1 1.5 2 2.5 3 3.5 4 mM Normally, 1.5mM MgCl 2 is optimal Best supplied as separate tube Always vortex thawed MgCl 2 Mg 2+ concentration will be affected by the amount of DNA, primers and nucleotides

51
USE MASTERMIXES WHERE POSSIBLE

52
How Powerful is PCR? PCR can amplify a usable amount of DNA (visible by gel electrophoresis) in ~2 hours. The template DNA need not be highly purified — a boiled bacterial colony. The PCR product can be digested with restriction enzymes, sequenced or cloned. PCR can amplify a single DNA molecule, e.g. from a single sperm.
 in ~2 hours. The template DNA need not be highly purified — a boiled bacterial colony. The PCR product can be digested with restriction enzymes, sequenced or cloned. PCR can amplify a single DNA molecule, e.g. from a single sperm..")
53
Applications of PCR Amplify specific DNA sequences (genomic DNA, cDNA, recombinant DNA, etc.) for analysis Introduce sequence changes at the ends of fragments Rapidly detect differences in DNA sequences (e.g., length) for identifying diseases or individuals Identify and isolate genes using degenerate oligonucleotide primers –Design mixture of primers to bind DNA encoding conserved protein motifs Genetic diagnosis - Mutation detection basis for many techniques to detect gene mutations (sequencing) - 1/6 X 10 -9 bp
 for analysis Introduce sequence changes at the ends of fragments Rapidly detect differences in DNA sequences (e.g., length) for identifying diseases or individuals Identify and isolate genes using degenerate oligonucleotide primers –Design mixture of primers to bind DNA encoding conserved protein motifs Genetic diagnosis - Mutation detection basis for many techniques to detect gene mutations (sequencing) - 1/6 X bp")
54
Paternity testing Mutagenesis to investigate protein function Quantify differences in gene expression Reverse transcription (RT)-PCR Identify changes in expression of unknown genes Differential display (DD)-PCR Forensic analysis at scene of crime Industrial quality control Applications of PCR
-PCR Identify changes in expression of unknown genes Differential display (DD)-PCR Forensic analysis at scene of crime Industrial quality control Applications of PCR")
56
Sequencing of DNA by the Sanger method

Similar presentations

>")


 fourth lecture Zoology department 2007 Dr.Maha H. Daghestani.>")
 Analysis of DNA (Sequencing) Chemical Synthesis.>")

 Analysis of DNA (Sequencing) Chemical Synthesis of DNA.>")

 Prepared by: M. Mohsin Ali Dynamo.>")





 Presented by: Afsaneh Bazgir Polymerase Chain Reaction>")

>")

 2. Restriction fragment length polymorphism (RFLP) 3. DNA sequencing.>")