Download presentation
Presentation is loading. Please wait.

1
Pairwise Sequence Alignment
2005/9/22 2005 Autumn / YM / Bioinformatics

2
Outline Motivation A simple alignment problem Scoring alignments
Substition matrices Gap penalties Dynamic programming Global vs. local alignment

3
Reference D.W. Mount / Bioinformatics / Ch.3 Slides from
Prof. C.H.Chang UW / Genomic Informatics / W.S. Noble

4
Motivation Why align two protein or DNA sequences?
Determine whether they are descended from a common ancestor (homologous). Infer a common function. Locate functional elements (motifs or domains). Infer protein structure, if the structure of one of the sequences is known.
. Infer a common function. Locate functional elements (motifs or domains). Infer protein structure, if the structure of one of the sequences is known.")
5
Example

6
Brief History Robinson (1938) described the 1st algorithm for Longest Common Subsequence problem Levenshtein (1966) introduced “edit distance” between strings Min. # of edit operations needed to transform from one string to another No algorithm described Gibbs & McIntyre (1970): dot matrix analysis Needleman & Wunsch (1970) “re-discovered” the algorithm for sequence alignment using DP Smith & Waterman (1981) proposed a clever modification of DP for local alignment
 introduced edit distance between strings. Min. # of edit operations needed to transform from one string to another. No algorithm described. Gibbs & McIntyre (1970): dot matrix analysis. Needleman & Wunsch (1970) re-discovered the algorithm for sequence alignment using DP. Smith & Waterman (1981) proposed a clever modification of DP for local alignment.")
7
Brief History Knuth (1973) suggested a filtering idea for pattern matching 高德纳 (Donald Knuth) Dumas & Ninio (1982) first applied the filtering idea for fast sequence comparison Lipman & Pearson (1985): FASTA Altschul et al. (1990): BLAST
 first applied the filtering idea for fast sequence comparison. Lipman & Pearson (1985): FASTA. Altschul et al. (1990): BLAST.")
8
Pairwise Alignment Methods
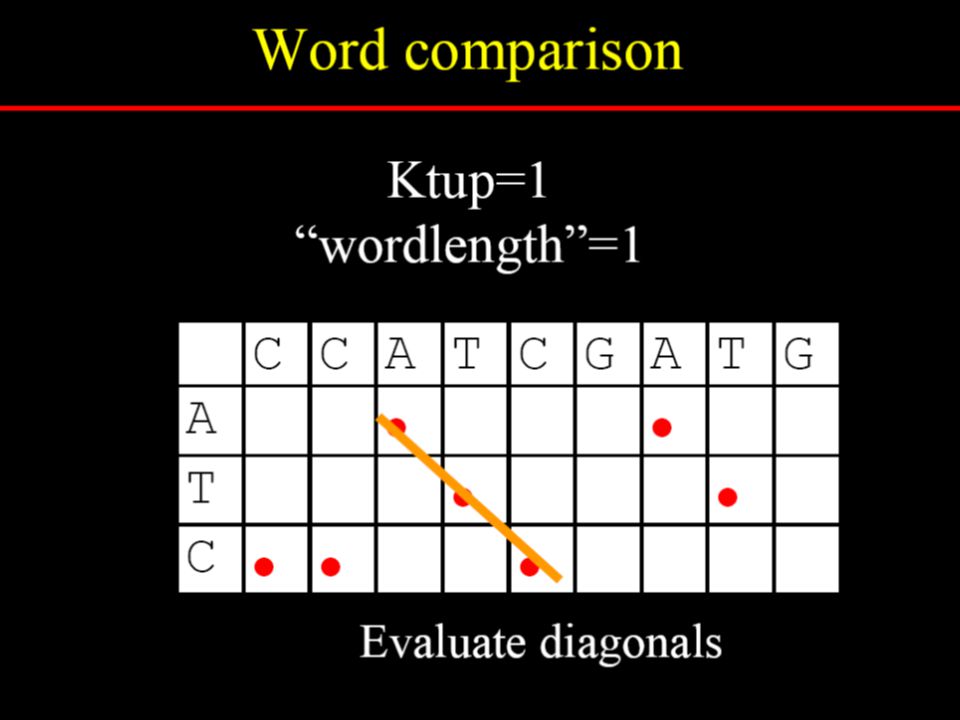
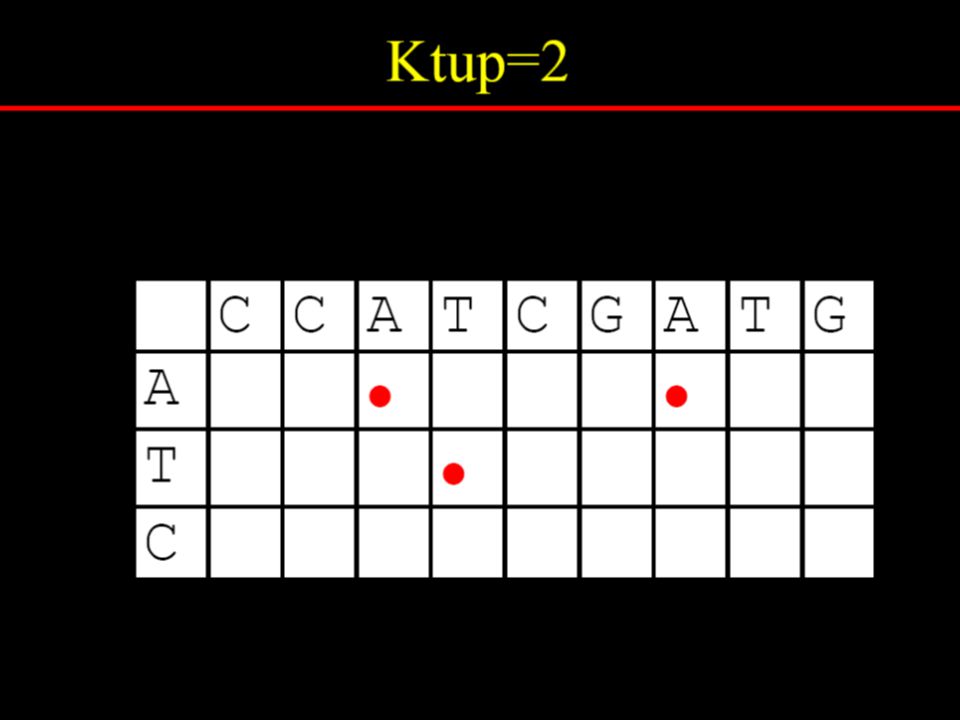
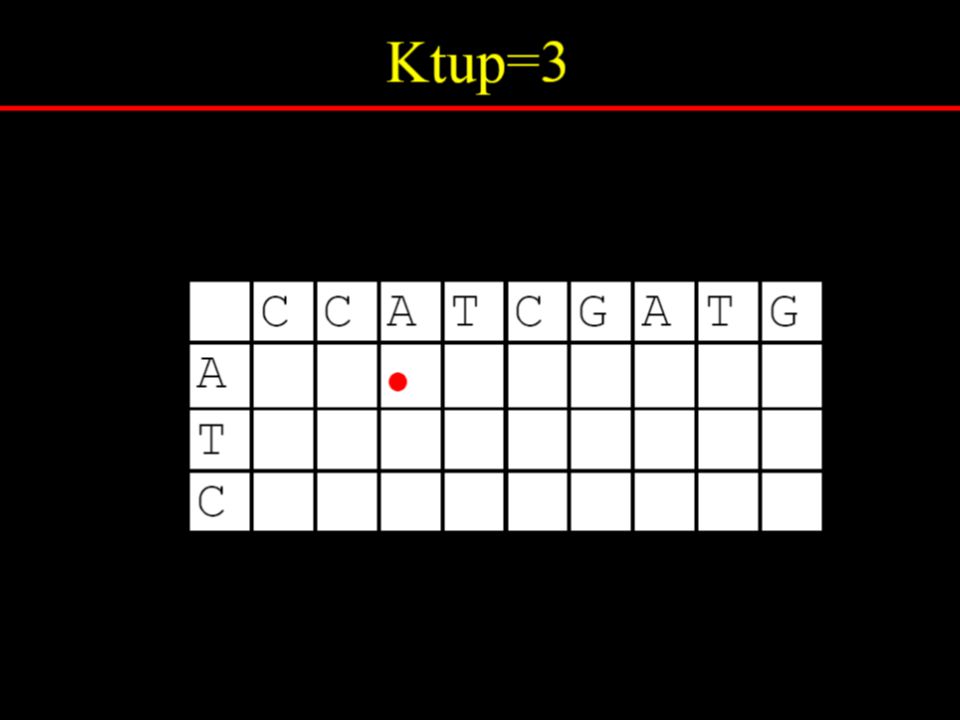
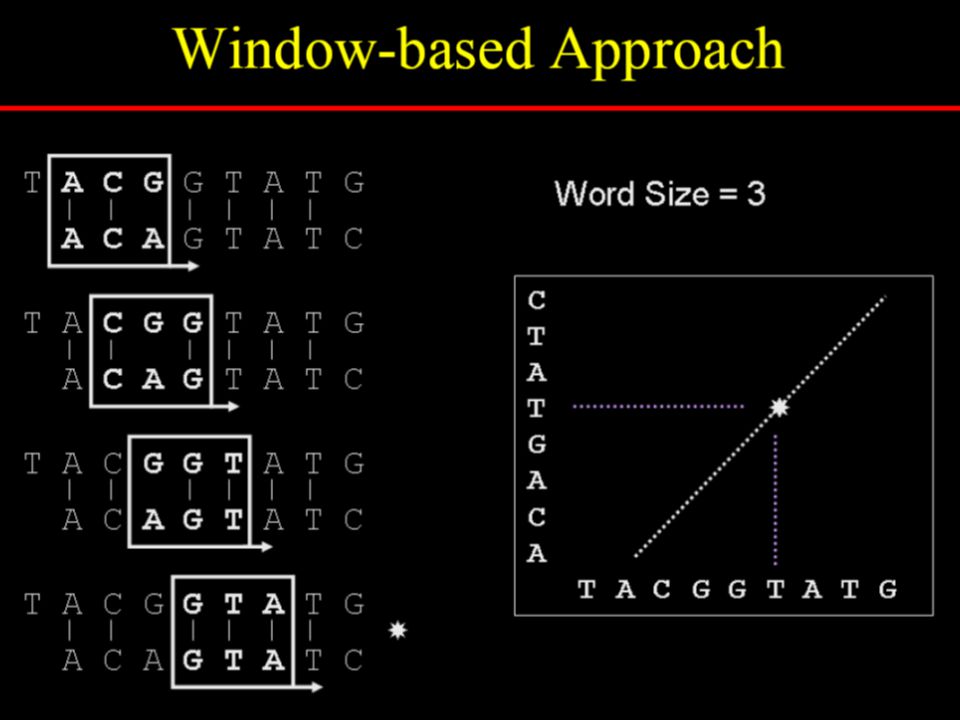
Dotplot analysis method Dynamic programming (DP) algorithm Word / k-tuple methods
 algorithm. Word / k-tuple methods.")
9
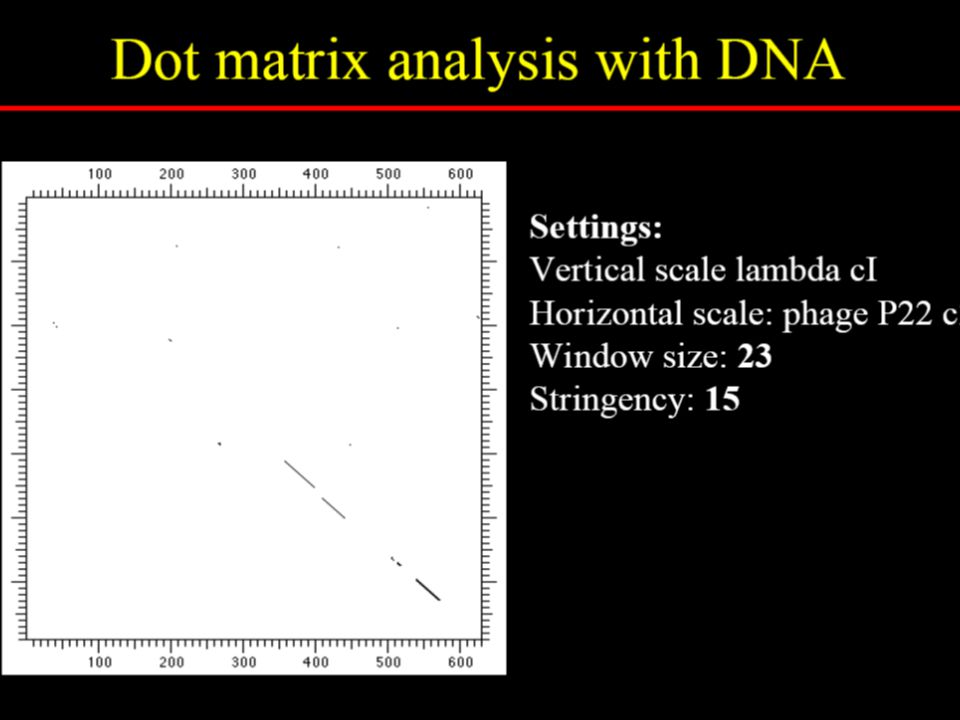
How dotplot analysis works

10
How dotplot analysis works
Direct Repeat Reversed Repeat Aligned sequence

11
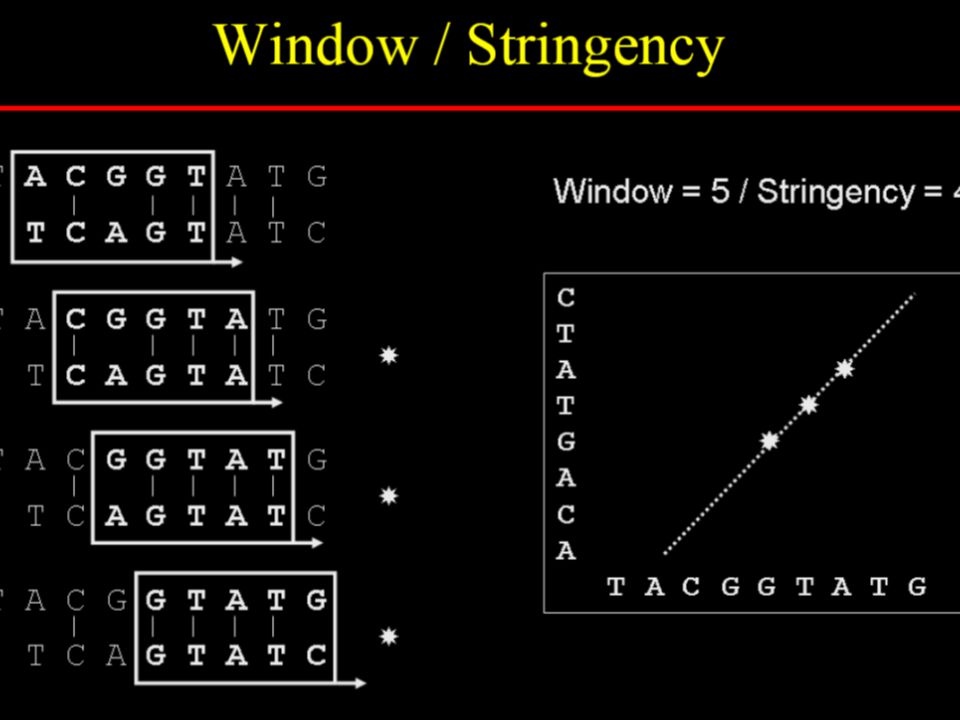
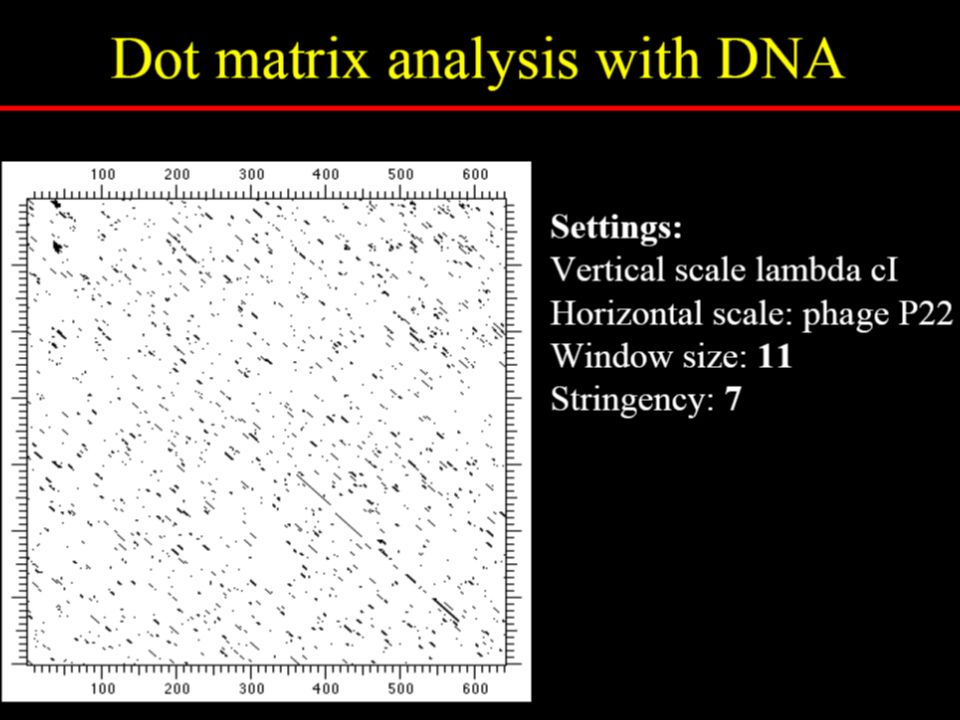
To reduce random noise in dotplots
Specify a window size, w Examine w residues at a time Count how many matches are among the w pairs of residues

20
Dotlet

21
A simple alignment problem.
Problem: find the best pairwise alignment of GAATC and CATAC.

22
Scoring alignments GAATC CATAC GAAT-C C-ATAC -GAAT-C C-A-TAC GAATC-
We need a way to measure the quality of a candidate alignment. Alignment scores consist of two parts: a substition matrix, and a gap penalty.

23
Scoring aligned bases Purine A G Pyrimidine C T Transversion
(low score) Transition (high score)
 Transition. (high score)")
24
Scoring aligned bases GAATC CATAC Purine A G Pyrimidine C T A C G T 10
Transversion Transition A hypothetical substitution matrix: GAATC CATAC A C G T 10 -5 = 5

25
Scoring aligned bases GAAT-C CA-TAC Purine A G Pyrimidine C T A C G T
Transversion (expensive) Transition (cheap) A hypothetical substitution matrix: GAAT-C CA-TAC A C G T 10 -5 ? ? + 10 = ?
 Transition. (cheap) A hypothetical substitution matrix: GAAT-C. CA-TAC. A. C. G. T =")
26
Scoring gaps Linear gap penalty: every gap receives a score of d.
Affine gap penalty: opening a gap receives a score of d; extending a gap receives a score of e. GAAT-C d=-4 CA-TAC = 17 G--AATC d=-4 CATA--C e=-1 = 5

27
A simple alignment problem.
Problem: find the best pairwise alignment of GAATC and CATAC. Use a linear gap penalty of -4. Use the following substitution matrix: A C G T 10 -5

28
How many possibilities?
GAATC CATAC GAAT-C C-ATAC -GAAT-C C-A-TAC GAATC- CA-TAC GAAT-C CA-TAC GA-ATC CATA-C How many different alignments of two sequences of length N exist?

29
How many possibilities?
GAATC CATAC GAAT-C C-ATAC -GAAT-C C-A-TAC GAATC- CA-TAC GAAT-C CA-TAC GA-ATC CATA-C How many different alignments of two sequences of length N exist? Too many to enumerate!

30
-G- CAT DP matrix G A T C The value in position (i,j) is the score of the best alignment of the first i positions of the first sequence versus the first j positions of the second sequence. -8
 is the score of the best alignment of the first i positions of the first sequence versus the first j positions of the second sequence. -8.")
31
DP matrix -8 G A T C -12 -G-A CAT-
Moving horizontally in the matrix introduces a gap in the sequence along the left edge.

32
DP matrix -8 G A T C -12 -G-- CATA
Moving vertically in the matrix introduces a gap in the sequence along the top edge.

33
Initialization G A T C

34
G - Introducing a gap G A T C -4

35
- C DP matrix G A T C -4

36
DP matrix G A T C -4 -8

37
G C DP matrix G A T C -4 -5

38
----- CATAC DP matrix G A T C -4 -8 -12 -16 -20 -5

39
DP matrix G A T C -4 -8 -12 -16 -20 -5 ?

40
DP matrix G A T C -4 -8 -12 -16 -20 -5 -G CA G- CA --G CA- -4 -9 -12
-4 -8 -12 -16 -20 -5 -4 -4
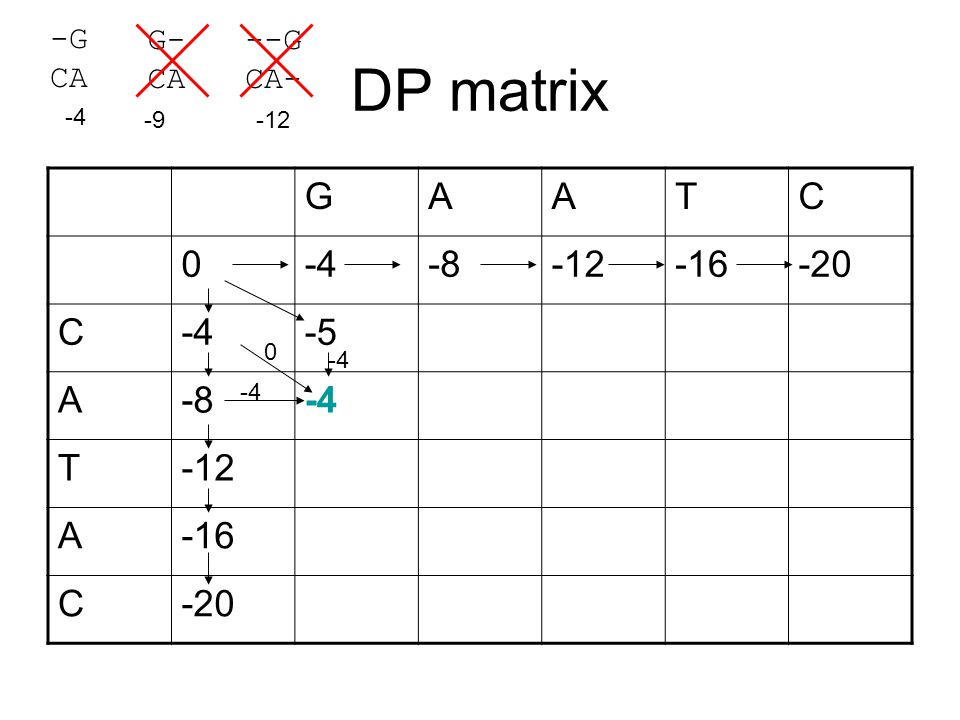
41
DP matrix G A T C -4 -8 -12 -16 -20 -5 ?

42
DP matrix G A T C -4 -8 -12 -16 -20 -5

43
DP matrix G A T C -4 -8 -12 -16 -20 -5 ?

44
What is the alignment associated with this entry?
DP matrix G A T C -4 -8 -12 -16 -20 -5 -9 5 1 2 -2 What is the alignment associated with this entry?

45
DP matrix G A T C -4 -8 -12 -16 -20 -5 -9 5 1 2 -2 -G-A CATA

46
Find the optimal alignment, and its score.
DP matrix G A T C -4 -8 -12 -16 -20 -5 -9 5 1 2 -2 ? Find the optimal alignment, and its score.

47
DP matrix G A T C -4 -8 -12 -16 -20 -5 -9 -13 -6 5 1 -3 -7 11 7 2 6 -2
-4 -8 -12 -16 -20 -5 -9 -13 -6 5 1 -3 -7 11 7 2 6 -2 17
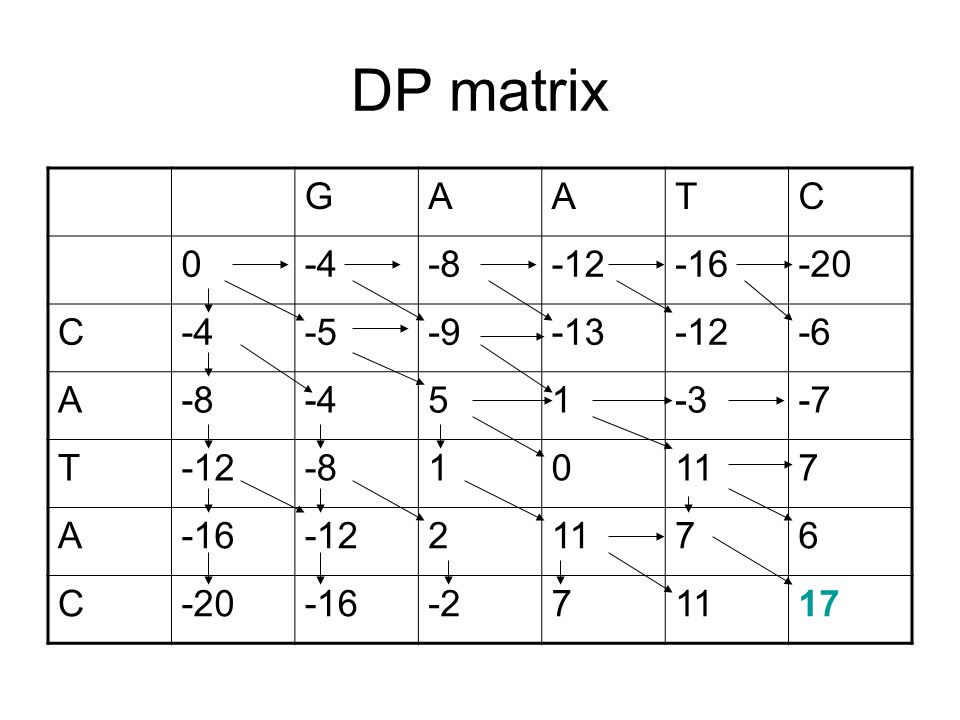
48
DP matrix GA-ATC CATA-C G A T C -4 -8 -12 -16 -20 -5 -9 -13 -6 5 1 -3
-4 -8 -12 -16 -20 -5 -9 -13 -6 5 1 -3 -7 11 7 2 6 -2 17
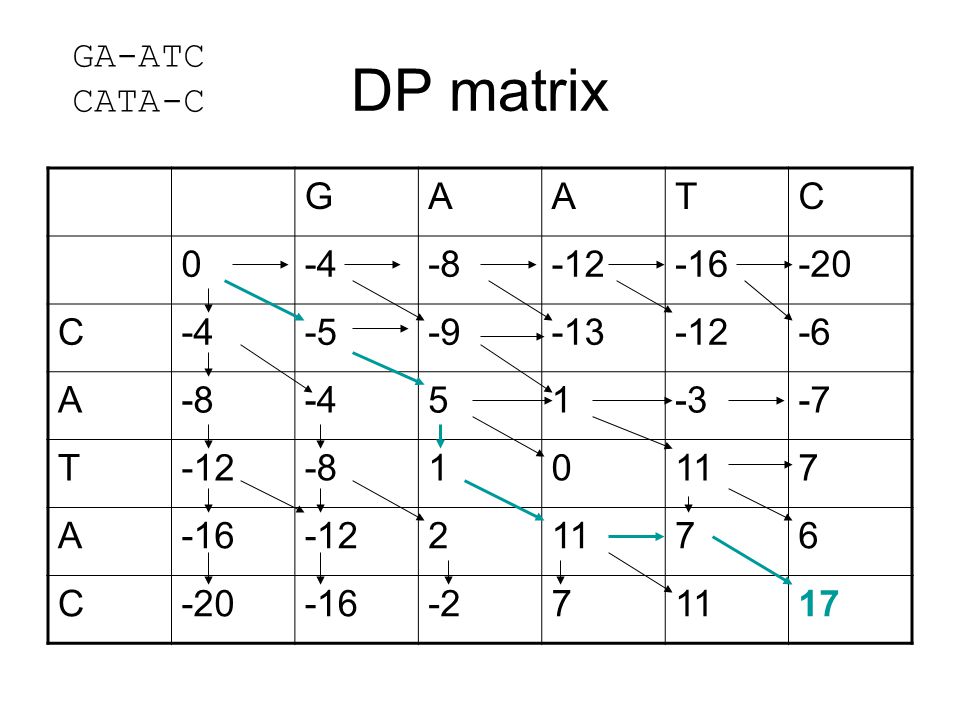
49
DP matrix GAAT-C CA-TAC G A T C -4 -8 -12 -16 -20 -5 -9 -13 -6 5 1 -3
-4 -8 -12 -16 -20 -5 -9 -13 -6 5 1 -3 -7 11 7 2 6 -2 17
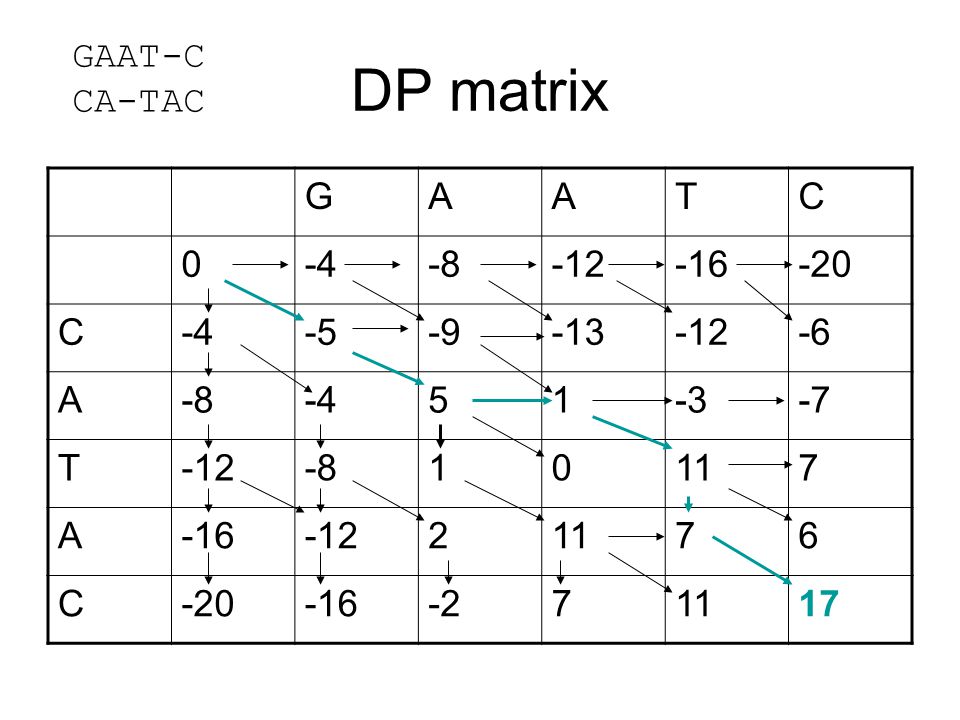
50
DP matrix GAAT-C C-ATAC G A T C -4 -8 -12 -16 -20 -5 -9 -13 -6 5 1 -3
-4 -8 -12 -16 -20 -5 -9 -13 -6 5 1 -3 -7 11 7 2 6 -2 17
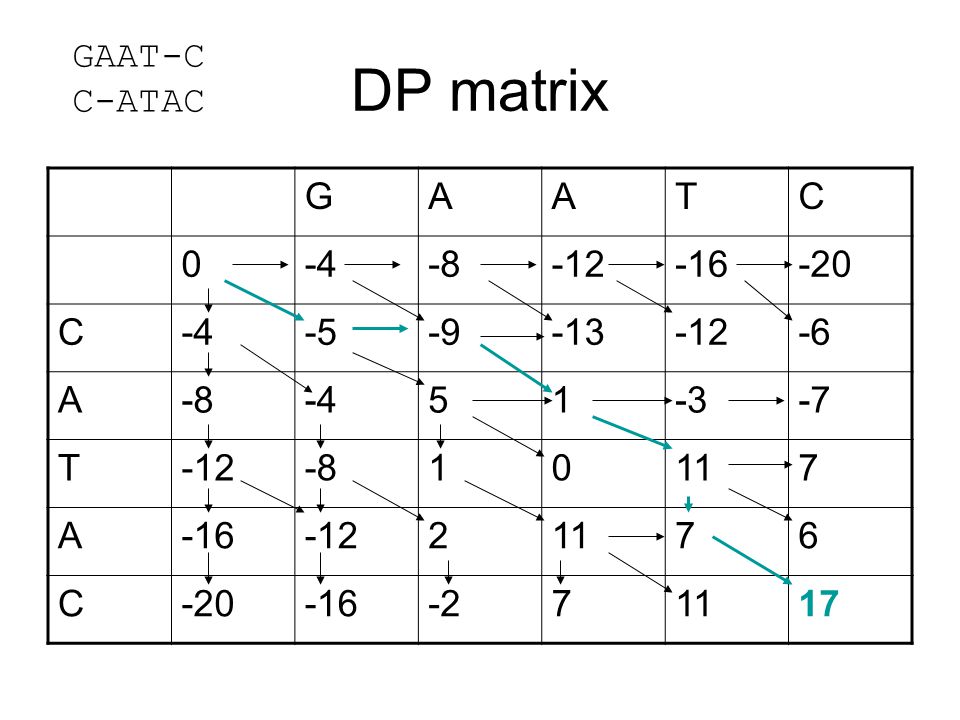
51
DP matrix GAAT-C -CATAC G A T C -4 -8 -12 -16 -20 -5 -9 -13 -6 5 1 -3
-4 -8 -12 -16 -20 -5 -9 -13 -6 5 1 -3 -7 11 7 2 6 -2 17
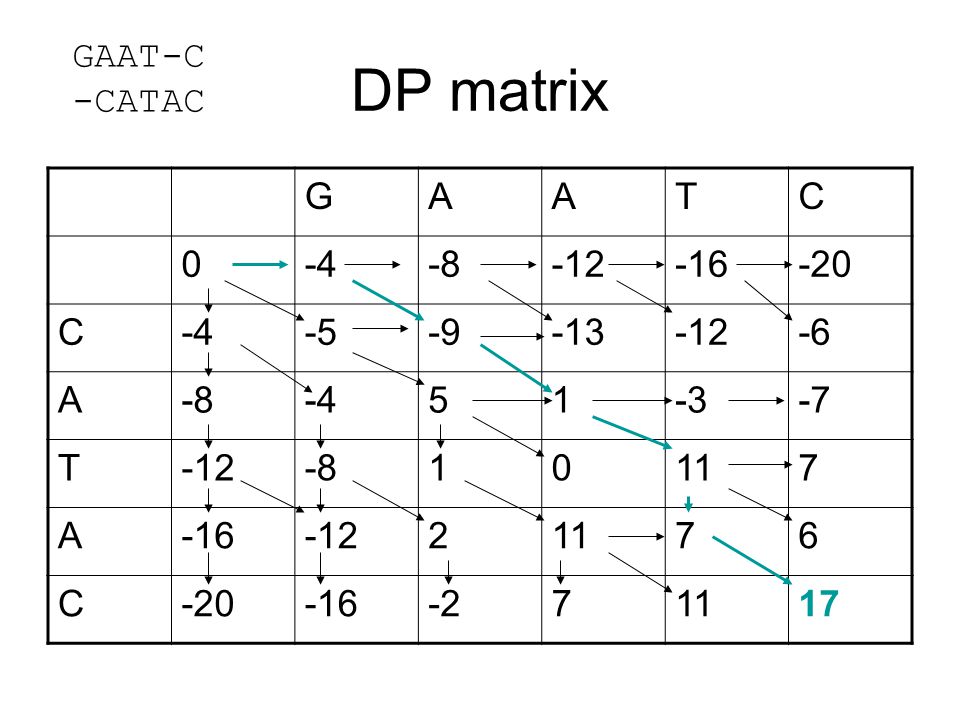
52
Multiple solutions GA-ATC CATA-C When a program returns a sequence alignment, it may not be the only best alignment. GAAT-C CA-TAC GAAT-C C-ATAC GAAT-C -CATAC

53
DP in equation form Align sequence x and y.
F is the DP matrix; s is the substitution matrix; d is the linear gap penalty.

54
DP in equation form

55
Dynamic programming Yes, it’s a weird name.
DP is closely related to recursion and to mathematical induction. We can prove that the resulting score is optimal.

56
Dynamic Programming A method for solving recursive problem
Break a problem into smaller subproblems Solve subproblems optimally, recursively Use these optimal solutions to construct an optimal solution for the original problem

57
Summary Scoring a pairwise alignment requires a substition matrix and gap penalties. Dynamic programming is an efficient algorithm for finding the optimal alignment. Entry (i,j) in the DP matrix stores the score of the best-scoring alignment up to those positions. DP iteratively fills in the matrix using a simple mathematical rule.
 in the DP matrix stores the score of the best-scoring alignment up to those positions. DP iteratively fills in the matrix using a simple mathematical rule.")
58
A simple exercise A G C Find the optimal alignment of AAG and AGC.
Use a gap penalty of d=-5. A C G T 2 -7 -5 A G C

59
A simple exercise A G C Find the optimal alignment of AAG and AGC.
Use a gap penalty of d=-5. A C G T 2 -7 -5 A G C

60
A simple exercise A G -5 -10 -15 C
Find the optimal alignment of AAG and AGC. Use a gap penalty of d=-5. A C G T 2 -7 -5 A G -5 -10 -15 C

61
A simple exercise A G -5 -10 -15 2 -3 -8 -1 C -6
Find the optimal alignment of AAG and AGC. Use a gap penalty of d=-5. A C G T 2 -7 -5 A G -5 -10 -15 2 -3 -8 -1 C -6

62
Traceback Start from the lower right corner and trace back to the upper left. Each arrow introduces one character at the end of each aligned sequence. A horizontal move puts a gap in the left sequence. A vertical move puts a gap in the top sequence. A diagonal move uses one character from each sequence.

63
A simple example A G -5 2 -3 -1 C -6
Find the optimal alignment of AAG and AGC. Use a gap penalty of d=-5. Start from the lower right corner and trace back to the upper left. Each arrow introduces one character at the end of each aligned sequence. A horizontal move puts a gap in the left sequence. A vertical move puts a gap in the top sequence. A diagonal move uses one character from each sequence. A G -5 2 -3 -1 C -6

64
A simple example A G -5 2 -3 -1 C -6
Find the optimal alignment of AAG and AGC. Use a gap penalty of d=-5. Start from the lower right corner and trace back to the upper left. Each arrow introduces one character at the end of each aligned sequence. A horizontal move puts a gap in the left sequence. A vertical move puts a gap in the top sequence. A diagonal move uses one character from each sequence. A G -5 2 -3 -1 C -6 AAG- AAG- -AGC A-GC

65
Local alignment A single-domain protein may be homologous to a region within a multi-domain protein. Usually, an alignment that spans the complete length of both sequences is not required.

66
BLAST allows local alignments
Global alignment Local alignment

67
Global alignment DP Align sequence x and y.
F is the DP matrix; s is the substitution matrix; d is the linear gap penalty.

68
Local alignment DP Align sequence x and y.
F is the DP matrix; s is the substitution matrix; d is the linear gap penalty.

69
Local DP in equation form

70
Local alignment Two differences with respect to global alignment:
No score is negative. Traceback begins at the highest score in the matrix and continues until you reach 0. Global alignment algorithm: Needleman-Wunsch. Local alignment algorithm: Smith-Waterman.

71
A simple example Find the optimal local alignment of AAG and AGC. Use a gap penalty of d=-5. A C G T 2 -7 -5 A G C

72
A simple example Find the optimal local alignment of AAG and AGC. Use a gap penalty of d=-5. A C G T 2 -7 -5 A G C

73
A simple example Find the optimal local alignment of AAG and AGC. Use a gap penalty of d=-5. A C G T 2 -7 -5 A G 2 4 C

74
A simple example Find the optimal local alignment of AAG and AGC. Use a gap penalty of d=-5. A C G T 2 -7 -5 A G 2 4 C AG

75
Local alignment Find the optimal local alignment of AAG and GAAGGC. Use a gap penalty of d=-5. A C G T 2 -7 -5 A G C

76
Local alignment Find the optimal local alignment of AAG and GAAGGC. Use a gap penalty of d=-5. A C G T 2 -7 -5 A G 2 4 6 1 C

77
Substitution matrices
Find the optimal local alignment of AAG and GAAGGC. Use a gap penalty of d=-5. A C G T 2 -7 -5 A G 2 4 6 1 C Where did this substitution matrix come from?

78
Nucleic acid PAM matrices
PAM = point accepted mutation 1 PAM = 1% probability of mutation at each sequence position. A uniform PAM1 matrix: A G T C 0.99

79
Transitions and transversions
Transitions (A G or C T) are more likely than transversions (A T or G C) Assume that transitions are three times as likely: A G T C 0.99 0.006 0.002
 are more likely than transversions (A T or G C) Assume that transitions are three times as likely: A. G. T. C")
80
Distant relatives If the probability of a substitution is 2%, simply multiply the probabilities from 1% by themselves. A PAM N matrix is computed by raising PAM 1 to the Nth power. A G T C

81
Better gap scoring Real gaps are often more than one letter long.
>gi|729942|sp|P40601|LIP1_PHOLU Lipase 1 precursor (Triacylglycerol lipase) Length = 645 Score = 33.5 bits (75), Expect = 5.9 Identities = 32/180 (17%), Positives = 70/180 (38%), Gaps = 9/180 (5%) Query: 2038 IYSLYGLYNVPYENLFVEAIASYSDNKIRSKSRRVIATTLETVGYQTANGKYKSESYTGQ 2097 +++ YGL+ Y Y D K +R N G+ Sbjct: 441 VFTAYGLWRY-YDKGWISGDLHYLDMKYEDITRGIVLNDW----LRKENASTSGHQWGGR 495 Query: 2098 LMAGYTYMMPENINLTPLAGLRYSTIKDKGYKETGTTYQNLTVKGKNYNTFDGLLGAKVS 2157 + AG P KGY+E+G Y++ G LG ++ Sbjct: 496 ITAGWDIPLTSAVTTSPIIQYAWDKSYVKGYRESGNNSTAMHFGEQRYDSQVGTLGWRLD 555 Query: 2158 SNINVNEIVLTPELYAMVDYAFKNKVSAIDARLQGMTAPLPTNSFKQSKTSFDVGVGVTA 2217 +N P F +K I S KQ G+ A Sbjct: 556 TNFG----YFNPYAEVRFNHQFGDKRYQIRSAINSTQTSFVSESQKQDTHWREYTIGMNA 611 Real gaps are often more than one letter long.
 Length = 645. Score = 33.5 bits (75), Expect = 5.9. Identities = 32/180 (17%), Positives = 70/180 (38%), Gaps = 9/180 (5%) Query: 2038 IYSLYGLYNVPYENLFVEAIASYSDNKIRSKSRRVIATTLETVGYQTANGKYKSESYTGQ YGL+ Y+ ++ Y D K +R ++ + N + G+ Sbjct: 441 VFTAYGLWRY-YDKGWISGDLHYLDMKYEDITRGIVLNDW----LRKENASTSGHQWGGR 495. Query: 2098 LMAGYTYMMPENINLTPLAGLRYSTIKDKGYKETGTTYQNLTVKGKNYNTFDGLLGAKVS AG P+ + KGY+E+G + + Y++ G LG ++ Sbjct: 496 ITAGWDIPLTSAVTTSPIIQYAWDKSYVKGYRESGNNSTAMHFGEQRYDSQVGTLGWRLD 555. Query: 2158 SNINVNEIVLTPELYAMVDYAFKNKVSAIDARLQGMTAPLPTNSFKQSKTSFDVGVGVTA N P ++ F +K I S KQ + +G+ A. Sbjct: 556 TNFG----YFNPYAEVRFNHQFGDKRYQIRSAINSTQTSFVSESQKQDTHWREYTIGMNA 611. Real gaps are often more than one letter long.")
82
Affine gap penalty LETVGY W----L -5 -1 -1 -1
Separate penalties for gap opening and gap extension. This requires modifying the DP algorithm to store three values in each box.

83
Summary Local alignment finds the best match between subsequences.
Smith-Waterman local alignment algorithm: No score is negative. Trace back from the largest score in the matrix. Substitution matrices represent the probability of mutations. Affine gap penalties include a large gap opening penalty and small gap extension penalty.

Similar presentations


















 The Mechanics of Alignments.>")

