Download presentation
Presentation is loading. Please wait.

1
Identification of an informative and accurate region of the HCV genome for phylogenetic analyses Patrik Medstrand patrik.medstrand@med.lu.se Clinical Virology Department of Laboratory Medicine Malmö Lund University, Sweden 11 th annual conference of new Visby network Vilnius, April 25-27, 2014

2
To investigate the dynamics of the HCV epidemic in the Baltic region using genetic and epidemiological data (date and site of sample collection, information of potential ”risk group”) To initiate a joint project among participants in the network (part of the proposal to SI) AIMS
 To initiate a joint project among participants in the network (part of the proposal to SI) AIMS")
3
Phylogenetics define genetic relationships We will study the genetic relationship among HCV strains that are circulating in the Baltic region. We could address: Relationship among HCV strains in the region (cities, countries, risk groups, general population) this can be done by “classical” phylogenetic studies, where groups (“clusters”) are defined
 this can be done by classical phylogenetic studies, where groups ( clusters ) are defined.")
4
Cluster definitions = need statistics To define relationships (clusters; epidemiological links defined by a common ancestral HCV strain) we need to use statistics. Limited information will obscure the identification of true relationships. Information or power in phylogenetic inference is represented by genetic information in terms of nucleotide sequences. This is similar to any statistical comparison - to small groups or limited information does not allow us to draw any robust conclusions.

5
More might be better 1 kb 9 kb Subset Complete

6
From phylogenetics to phylodynamics Unrooted Genetic relationshipRooted and with time estimates Colors: Geographic locations Risk groups etc NEW INSIGHTS

7
Using phylodynamic analysis, we investigate the extent the HCV epidemic in three metropolitan areas of Sweden were linked or separate. We found evidence for one early introduction (Western Europe to Gothenburg in 1958; panel A) and rapid dissemination (from Gothenburg to Stockholm and Malmö 1965- 1968; panels B-C), whereas the later epidemic (after 1975) were characterized by HCV strains that were introduced from regions outside Sweden (Western Europe and USA; panel D), indicating limited epidemic links within Sweden during this later time period (Jerkeman et al, manuscript in preparation). Panel E: exponential growth from ~1960-1980. A. B. C. D. Phylodynamics can inform about migrations and growth of the epidemic historical and more recent Similar studies can be performed on other “traits”, such as risk groups E 1960 1965 1968 1980
 and rapid dissemination (from Gothenburg to Stockholm and Malmö ; panels B-C), whereas the later epidemic (after 1975) were characterized by HCV strains that were introduced from regions outside Sweden (Western Europe and USA; panel D), indicating limited epidemic links within Sweden during this later time period (Jerkeman et al, manuscript in preparation). Panel E: exponential growth from ~ A. B. C. D. Phylodynamics can inform about migrations and growth of the epidemic historical and more recent Similar studies can be performed on other traits , such as risk groups E")
8
Goal of present study 1. Identify phylogenetically informative genome regions that Allow identification of a reasonable number and correct clusters Allow reconstruction of the “true” phylogeny in comparison to the phylogeny reconstructed from near full-length HCV genomes 2. Establish a convenient PCR and sequencing protocol

9
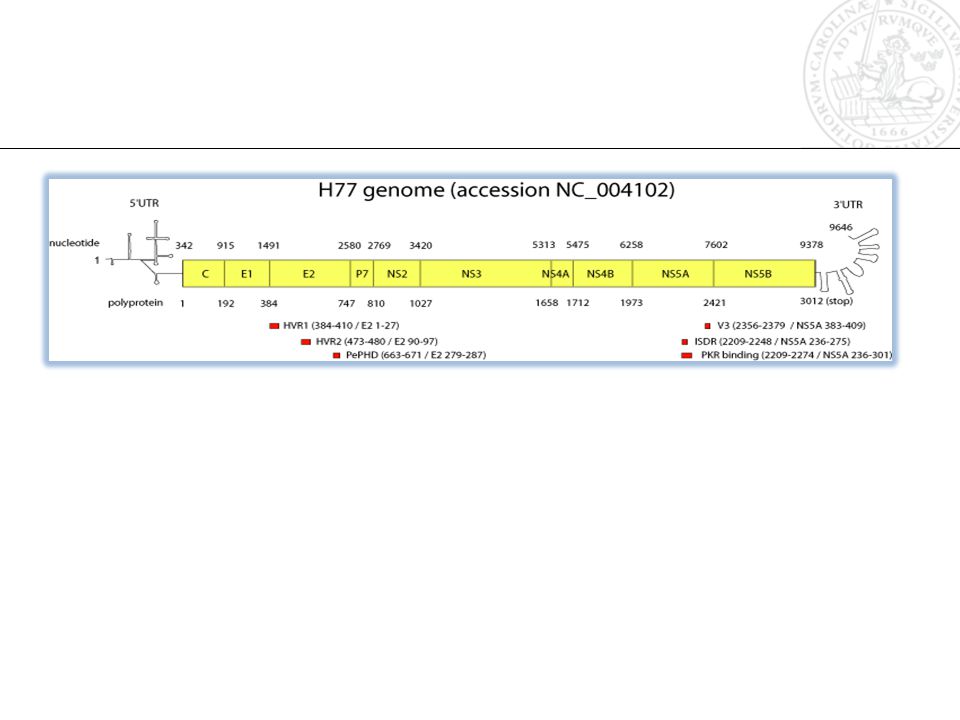
Genome regions Sequence similarity Number of sequences (country and year info) E1-E2 P7 P7-NS2 NS5A NS5B NS5A-NS5B NS5Bsh
 E1-E2 P7 P7-NS2 NS5A NS5B NS5A-NS5B NS5Bsh")
10
Data set and Methods 143 near full length HCV 1a genomes (polyprotein region) were obtained from the Los Alamos HCV database The data set was used to create 7 subsets representing 7 subgenomic regions ML trees were constructed using Garli v2.0 using GTR+I+G subst model Branch support was estimated using the Shimodaira-Hasegawa (SH) test as implemented in PhyML False positive branches were defined as branches with statistical support (SH > 0.9) in ML-trees of subgenomic regions, that were absent in the ML-tree obtained from the polyprotein region (“true” tree) Accuracy (topology-testing) of phylogenies obtained from subgenomic regions were inferred by statistical comparison to the “true” tree using the SH-test implemented in TreePuzzle and Consel
 were obtained from the Los Alamos HCV database The data set was used to create 7 subsets representing 7 subgenomic regions ML trees were constructed using Garli v2.0 using GTR+I+G subst model Branch support was estimated using the Shimodaira-Hasegawa (SH) test as implemented in PhyML False positive branches were defined as branches with statistical support (SH > 0.9) in ML-trees of subgenomic regions, that were absent in the ML-tree obtained from the polyprotein region ( true tree) Accuracy (topology-testing) of phylogenies obtained from subgenomic regions were inferred by statistical comparison to the true tree using the SH-test implemented in TreePuzzle and Consel")
11
Branch support Polyprotein E1-E2 P7 P7-NS2 NS5A-NS5B NS5A NS5B NS5B-sh (9036 bp) (1236 bp) (933 bp) (1455 bp) (2934 bp) (1272 bp) (1688 bp) (640 bp) 75 28 18 22 35 26 28 8 Supported Branches (N) FP (%) True supported Branches (N) 75 1 - 39 23 22 37 15 46 65 75 17 14 17 22 22 15 3
 (1236 bp) (933 bp) (1455 bp) (2934 bp) (1272 bp) (1688 bp) (640 bp) Supported Branches (N) FP (%) True supported Branches (N)")
12
Topology support E1-E2 P7 P7-NS2 NS5A-NS5B NS5A NS5B NS5B-sh (1236 bp) (933 bp) (1455 bp) (2934 bp) (1272 bp) (1688 bp) (640 bp) 27 35 38 37 39 21 12 Branches in subgenomic tree supported in true tree (N) Topology difference of sub- genomic and true tree (p-value) - <0.01 <0.01 <0.01 0.06 0.06 <0.01 <0.01
 (933 bp) (1455 bp) (2934 bp) (1272 bp) (1688 bp) (640 bp) Branches in subgenomic tree supported in true tree (N) Topology difference of sub- genomic and true tree (p-value) - <0.01 <0.01 < <0.01 <0.01")
13
Conclusions The 1272-bp region of NS5A displayed the lowest FP-rate compared to other subgenomic regions analyzed The NS5A and NS5A-NS5B trees conformed topologies of the true tree. In total, 39 NS5A branches of a total 75 branches were shared with the true tree. Among those, 22 branches had statistical support. The NS5A region represents a trade-off between phylogenetic accuracy/information in comparison to full-length genome sequencing, and may be suitable for phylogenetic and phylodynamics studies of HCV The preliminary findings shown here will be confirmed using other HCV subgenotypes and methods PCR protocols will be established and shared to network members

14
Lund University, Sweden Anders Widell Per Björkman Anna Jerkeman Marianne Alanko Vilma Molnegren Joakim Esbjörnsson HCV study ACKNOWLEDGEMENT Thanks to Anders and Joakim for presenting this! To bad that I couldn’t come to Vilnius but are looking forwards to see you all soon!

16
Alternative regions Sequence similarity Number of sequences (country and year info) E1-E2 P7 P7-NS2 NS5A NS5B NS5A-NS5B NS5Bsh
 E1-E2 P7 P7-NS2 NS5A NS5B NS5A-NS5B NS5Bsh")
Similar presentations









>")









