Download presentation
Presentation is loading. Please wait.

1
Recently Issued OHRP Documents: Guidance on Subject Withdrawal and Draft Revised FWA Secretary’s Advisory Committee on Human Research Protections October 20, 2010 Michael A. Carome, M.D. CAPT, U.S. Public Health Service Associate Director for Regulatory Affairs Office for Human Research Protections

2
Overview Guidance on Withdrawal of Subjects from Research: Data Retention and Other Related Issues – Historical background – Key content – Changes in comparison to draft guidance issued for public comment Draft revised Federalwide Assurance (FWA) documents – Historical background – Proposed changes in comparison to current FWA – How to submit and view comments
 documents – Historical background – Proposed changes in comparison to current FWA – How to submit and view comments")
3
Guidance on Withdrawal of Subjects from Research: Data Retention and Other Related Issues

4
Guidance on Subject Withdrawal: Historical Background The draft document, Guidance on Important Considerations for When Participation of Human Subjects in Research is Discontinued, was issued for public comment on December 1, 2008. The comment period closed January 30, 2009. (See http://www.hhs.gov/ohrp/requests/com120108.html) On December 1, 2008, FDA also issued its Guidance for Sponsors, Clinical Investigators, and IRBs: Data Retention When Subjects Withdraw from FDA-Regulated Clinical Trials. 20 individuals, 2 Federal agencies, and 8 private organizations submitted comments to OHRP.

5
Guidance on Subject Withdrawal: Key Content What does it mean when a subject withdraws from a research study? May an investigator retain and analyze already collected data about a subject who withdraws from the research or whose participation is terminated by the investigator? Can investigators honor subjects’ requests to have their data destroyed or excluded from any analysis? Should the withdrawal of a subject from a research study be documented? What is the relationship of this guidance to FDA’s guidance on this issue and to the HIPAA Privacy Rule? When seeking the informed consent of subjects, what should investigators tell subjects about data retention in the event the subjects withdraw?

6
Guidance on Subject Withdrawal: Key Content (cont.) The guidance clarifies that when a subject chooses to withdraw from (i.e., discontinue his or her participation in) an ongoing research study, or when an investigator terminates a subject’s participation in such a research study without regard to the subject’s consent, the investigator may retain and analyze already collected data relating to that subject, even if that data includes identifiable private information about the subject.
 The guidance clarifies that when a subject chooses to withdraw from (i.e., discontinue his or her participation in) an ongoing research study, or when an investigator terminates a subject’s participation in such a research study without regard to the subject’s consent, the investigator may retain and analyze already collected data relating to that subject, even if that data includes identifiable private information about the subject.")
7
Guidance on Subject Withdrawal: Changes in Comparison to the Draft Guidance Title changed All content regarding biospecimens that was included in the draft guidance document was removed from the final guidance document. The final guidance document includes more examples of social and behavioral research activities in order to emphasize that the guidance applies to such research, in addition to its applicability to biomedical research.

8
Guidance on Subject Withdrawal: Changes in Comparison to the Draft Guidance (cont.) The final guidance includes a recommendation that investigators plan for the possibility that subjects will withdraw from research and that they include a discussion of what withdrawal will mean and how it will be handled in their research protocols and informed consent documents. Furthermore, the final guidance addresses the question of what investigators, when seeking the informed consent of subjects, should tell the subjects about data retention in the event the subjects withdraw.

9
Draft Revised Federalwide Assurance (FWA)
")
10
Draft Revised FWA: Historical Background First version of the FWA issued in December 2000. Second version issued in March 2002. Initial electronic submission system implemented in December 2002. Current version of the FWA was issued in January 2005. As of December 31, 2005, this is the only assurance FWA accepts. Currently, there are separate FWA forms and Terms of Assurance for U.S. and non-U.S. institutions.

11
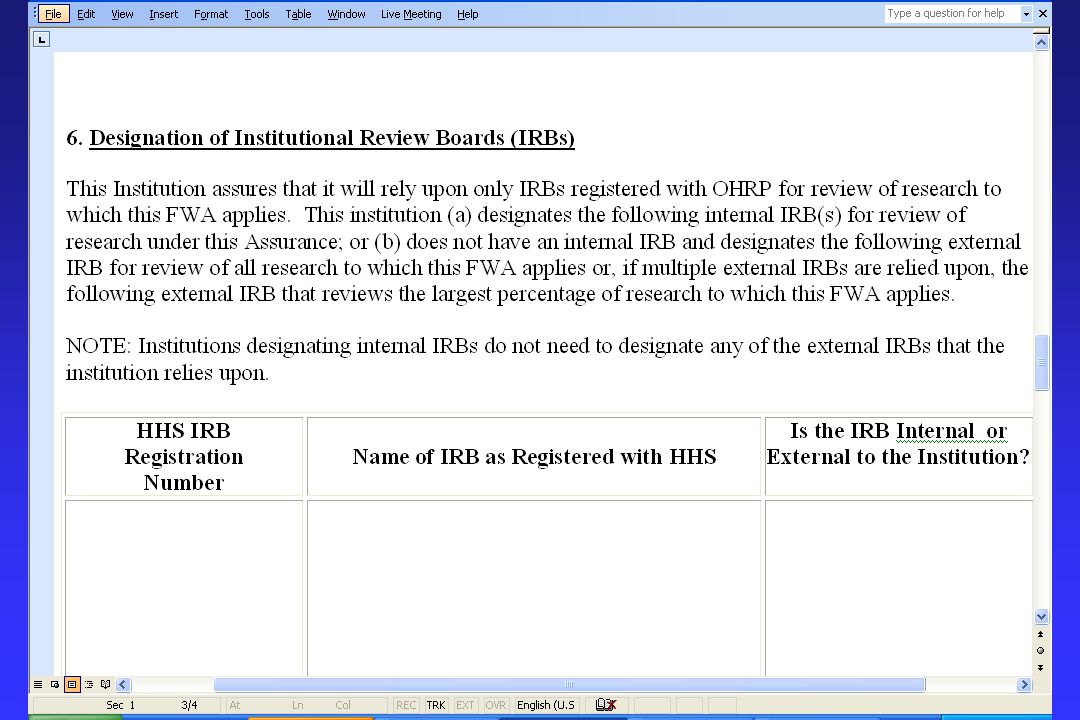
Draft Revised FWA: Proposed Changes The current separate FWA forms for U.S. and non-U.S. institutions have been combined into a single form. The Terms of Assurance document has been shortened and simplified. The current requirement that all IRBs (both internal and external IRBs) relied upon by the institution be specifically designated would be replaced with the requirement that only internal IRBs be specifically designated or that, if an institution does not have an internal IRB, only one external IRB be specifically designated.
 relied upon by the institution be specifically designated would be replaced with the requirement that only internal IRBs be specifically designated or that, if an institution does not have an internal IRB, only one external IRB be specifically designated..")
12
Draft Revised FWA: Proposed Changes (cont.) The revised FWA form would no longer request submission of the HHS Institution Profile code or the Federal Entity Identification number. The revised FWA form would allow the FWA to be signed by the institution’s signatory official electronically and eliminate the need for submission of a hard-copy signature page by mail or facsimile. The standard period of approval for an FWA would be increased from the current 3-year period to a 5- year period.

18
Draft Revised FWA: Deleted Terms 5. Scope of IRB(s)’s Responsibilities All human subjects research to which the FWA applies, except for research exempted or waived in accordance with Sections 101(b) or 101(i) of the Common Rule, will be reviewed, prospectively approved, and subject to continuing review at least annually by the designated IRB(s). The IRB(s) will have authority to approve, require modifications in, or disapprove the covered human subjects research. For research approved by the IRB(s), further appropriate review and approval by any department or agency conducting or supporting the research or by officials of the institution holding the FWA may be required.
’s Responsibilities All human subjects research to which the FWA applies, except for research exempted or waived in accordance with Sections 101(b) or 101(i) of the Common Rule, will be reviewed, prospectively approved, and subject to continuing review at least annually by the designated IRB(s). The IRB(s) will have authority to approve, require modifications in, or disapprove the covered human subjects research. For research approved by the IRB(s), further appropriate review and approval by any department or agency conducting or supporting the research or by officials of the institution holding the FWA may be required..")
19
Draft Revised FWA: Deleted Terms (cont.) 6. Informed Consent Requirements Except for research exempted or waived in accordance with Sections 101(b) or 101(i) of the Common Rule, informed consent for research to which the FWA applies will be: a) sought from each prospective subject or the subject's legally authorized representative, in accordance with, and to the extent required by, Section 116 of the Common Rule; and b) appropriately documented, in accordance with, and to the extent required by, Section 117 of the Common Rule.
 or 101(i) of the Common Rule, informed consent for research to which the FWA applies will be: a) sought from each prospective subject or the subject s legally authorized representative, in accordance with, and to the extent required by, Section 116 of the Common Rule; and b) appropriately documented, in accordance with, and to the extent required by, Section 117 of the Common Rule..")
20
Draft Revised FWA: Deleted Terms (cont.) 7. Requirement for Assurances for Collaborating Institutions When the Institution holding the FWA is either a) the primary awardee under a federal grant, contract, or cooperative agreement supporting research to which the FWA applies, or b) the coordinating center for federally- conducted or –supported research to which the FWA applies, the Institution is responsible for ensuring that all collaborating institutions engaged in such research operate under an appropriate OHRP-approved or other federally-approved assurance for the protection of human subjects....
 the primary awardee under a federal grant, contract, or cooperative agreement supporting research to which the FWA applies, or b) the coordinating center for federally- conducted or –supported research to which the FWA applies, the Institution is responsible for ensuring that all collaborating institutions engaged in such research operate under an appropriate OHRP-approved or other federally-approved assurance for the protection of human subjects.....")
21
Draft Revised FWA: Deleted Terms (cont.) 8. Written Agreements with Independent Investigators Who are not Otherwise Affiliated with the Institution When the Institution holding the FWA is either a) the primary awardee under a federal grant, contract, or cooperative agreement supporting research to which the FWA applies, or b) the coordinating center for federally- conducted or –supported research to which the FWA applies, the Institution is responsible for ensuring that all collaborating independent investigators engaged in such research operate under an appropriate OHRP-approved or other federally-approved assurance for the protection of human subjects….
 the primary awardee under a federal grant, contract, or cooperative agreement supporting research to which the FWA applies, or b) the coordinating center for federally- conducted or –supported research to which the FWA applies, the Institution is responsible for ensuring that all collaborating independent investigators engaged in such research operate under an appropriate OHRP-approved or other federally-approved assurance for the protection of human subjects…..")
22
Draft Revised FWA: Deleted Terms (cont.) 10. Compliance with the Terms of Assurance The Institution accepts and will follow items 1-9 above and is responsible for ensuring that (a) the IRB(s) designated under the FWA agree to comply with these terms; and (b) the IRB(s) possess appropriate knowledge of the local research context for all research to which the FWA applies (please refer to the OHRP Guidance on IRB Knowledge of Local Research Context on the OHRP website at http://www.hhs.gov/ohrp/humansubjects/guidance/loc al.htm).
 the IRB(s) designated under the FWA agree to comply with these terms; and (b) the IRB(s) possess appropriate knowledge of the local research context for all research to which the FWA applies (please refer to the OHRP Guidance on IRB Knowledge of Local Research Context on the OHRP website at al.htm)..")
23
Draft Revised FWA: Deleted Terms (cont.) 11. Assurance Training The OHRP Assurance Training Modules…describe the major responsibilities of the Institutional Signatory Official, the Human Protection Administrator (e.g., Human Subjects Administrator or Human Subjects Contact Person), and the IRB Chair(s) that must be fulfilled under the FWA. OHRP strongly recommends that the Institutional Signatory Official, the Human Protections Administrator, and the IRB Chair(s) personally complete the relevant OHRP Assurance Training Modules, or comparable training that includes the content of these modules, prior to submitting the FWA.
, and the IRB Chair(s) that must be fulfilled under the FWA. OHRP strongly recommends that the Institutional Signatory Official, the Human Protections Administrator, and the IRB Chair(s) personally complete the relevant OHRP Assurance Training Modules, or comparable training that includes the content of these modules, prior to submitting the FWA..")
24
Draft Revised FWA: Deleted Terms (cont.) 12. Educational Training OHRP strongly recommends that the Institution and the designated IRB(s) establish educational training and oversight mechanisms (appropriate to the nature and volume of its research) to ensure that research investigators, IRB members and staff, and other appropriate personnel maintain continuing knowledge of, and comply with, the following: relevant ethical principles; relevant federal regulations; …. Furthermore, OHRP recommends that a) IRB members and staff complete relevant educational training before reviewing human subjects research; and b) research investigators complete appropriate institutional educational training before conducting human subjects research.
 establish educational training and oversight mechanisms (appropriate to the nature and volume of its research) to ensure that research investigators, IRB members and staff, and other appropriate personnel maintain continuing knowledge of, and comply with, the following: relevant ethical principles; relevant federal regulations; …. Furthermore, OHRP recommends that a) IRB members and staff complete relevant educational training before reviewing human subjects research; and b) research investigators complete appropriate institutional educational training before conducting human subjects research..")
25
Draft Revised FWA: Submitting and Viewing Comments See OHRP website at http://www.hhs.gov/ohrp/requests/ for links to the draft documents and instructions for submitting comments. Docket can be found at www.regulations.gov. E-mail alerts when docket modified. Comment period closes October 25.

26
HHS-OPHS-2010-0023

Similar presentations




 is a review committee established to help protect the rights and welfare of human research subjects.>")

 U.S. Army Medical Research and Materiel Command (USAMRMC) Fort Detrick,>")















 Human Subject Research Office (HSRO) University of Miami and Affiliated Institutions.>")
