Download presentation
Presentation is loading. Please wait.

1
From sequence data to genomic prediction

2
Course overview Day 1 Day 2 Day 3 Day 4 & 5 Introduction
Generation, quality control, alignment of sequence data Detection of variants, quality control and filtering Day 2 Imputation from SNP array genotypes to sequence data Day 3 Genome wide association studies with SNP array and sequence variant genotypes Day 4 & 5 Genomic prediction with SNP array and sequence variant genotypes (BLUP and Bayesian methods) Use of genomic selection in breeding programs
 Use of genomic selection in breeding programs.")
3
Imputation Why impute? Approaches for imputation
Factors affecting accuracy of imputation Does imputation give you more power? Imputation to whole genome sequence variant genotypes

4
Why impute? Fill in missing genotypes from the lab
Merge data sets with genotypes on different arrays Eg. Affy and Illumina data Impute from low density to high density 7K-> 50K (save $$$) 50K->800K capture power of higher density? Better persistence of accuracy Sequence expensive, can we impute to full sequence data?
 50K->800K. capture power of higher density Better persistence of accuracy. Sequence expensive, can we impute to full sequence data")
5
Core concept Identity by state (IBS) Identity by descent (IBD)
A pair of individuals have the same allele at a locus Identity by descent (IBD) A pair of individuals have the same alleles at a locus and it traces to a common ancestor Imputation methods determine whether a chromosome segment is IBD
 A pair of individuals have the same alleles at a locus and it traces to a common ancestor. Imputation methods determine whether a chromosome segment is IBD.")
6
Causes of LD A chunk of ancestral chromosome is conserved in the current population Marker Haplotype 1 1 1 2

7
Core concept 2 Any individuals in a population may share a proportion of their genome identical by descent (IBD) IBD segments are the same and have originated in a common ancestor The closer the relationship the longer the IBD segments Pedigree relationships

8
Several methods for imputation
Two main categories: Family based Population based Or combination of the two Some of the most effective are Beagle (Browning and Browning, 2009), MACH (Li et al., 2010), Impute2 (Howie et al., 2009), AlphaPhase (Hickey et al 2011)
, MACH (Li et al., 2010), Impute2 (Howie et al., 2009), AlphaPhase (Hickey et al 2011)")
9
Several methods for imputation
Two main categories: Family based Population based Or combination of the two Some of the most effective are Beagle (Browning and Browning, 2009), MACH (Li et al., 2010), Impute2 (Howie et al., 2009), AlphaPhase (Hickey et al 2011)
, MACH (Li et al., 2010), Impute2 (Howie et al., 2009), AlphaPhase (Hickey et al 2011)")
10
Finding an IBD segment Sire 2 Progeny ? 2

11
Sire 2 IBD segment Progeny ? 2

12
Sire 2 Progeny 2 ?

13
Several methods for imputation
Two main categories: Family based Population based (exploits LD) Or combination of the two Some of the most effective are Beagle (Browning and Browning, 2009), MACH (Li et al., 2010), Impute2 (Howie et al., 2009), AlphaPhase (Hickey et al 2011)
 Or combination of the two. Some of the most effective are Beagle (Browning and Browning, 2009), MACH (Li et al., 2010), Impute2 (Howie et al., 2009), AlphaPhase (Hickey et al 2011)")
14
Population based imputation
Hidden Markov Models Has “hidden states” For target individuals these are “map” of reference haplotypes that have been inherited Imputation problem is to derive genotype probabilities given hidden states, sparse genotypes, recombination rates, other population parameters

15
Population based imputation
Reference population Target population Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet :

16
Population based imputation
Consider three markers, 4 reference haplotypes 0 1 1 0 1 0 1 0 1 0 0 1 Imputation?

17
Li and Stephens

18
Beagle

19
Imputation accuracy Accuracy = correlation of real and imputed genotypes Concordance = percentage (%) of genotypes called correctly
 of genotypes called correctly.")
20
Imputation accuracy Depends on Size of reference set
bigger the better! Density of markers extent of LD, effective population size Frequency of SNP alleles Genetic relationship to reference

21
Table 6. Accuracy of imputation from BovineLD genotypes to BovineSNP50 genotypes for Australian, French, and North American breeds. Boichard D, Chung H, Dassonneville R, David X, et al. (2012) Design of a Bovine Low-Density SNP Array Optimized for Imputation. PLoS ONE 7(3): e doi: /journal.pone
 Design of a Bovine Low-Density SNP Array Optimized for Imputation. PLoS ONE 7(3): e doi: /journal.pone")
22
Imputation accuracy Density of markers (extent of LD)
In Holstein Dairy cattle 3K -> 50K accuracy 0.93 7K -> 50K accuracy 0.98

23
Illumina Bovine HD array
We genotyped 898 Holstein heifers 47 Holstein Key ancestor bulls After (stringent) QC 634,307 SNPs
 QC 634,307 SNPs.")
24
Imputation 50K -> 800K Holsteins

25
Imputation accuracy Rare alleles?

26
Imputation accuracy Relationship to reference?

27
Imputation accuracy Effect of map errors?

28
Why more power with imputation
High accuracies of imputation demonstrate that we can infer haplotypes of animal genotyped with e.g. 3K accurately But potentially large number of haplotypes With imputed data can test single snp, only use 1 degree of freedom, rather than number of haplotypes

29
Why more power with imputation
Weigel et al. (2010)
")
30
Imputation Why impute? Approaches for imputation
Factors affecting accuracy of imputation Does imputation give you more power? Imputation to whole genome sequence variant genotypes

31
Which individuals to sequence?
Those which capture greatest genetic diversity? Select set of individuals which are likely to capture highest proportion of unique chromosome segments

32
Which individuals to sequence?
Let total number of individuals in population be n, number of individuals that can be sequenced be m. A = average relationship matrix among n individuals, from pedigree

33
An example A matrix…….. Pedigree Animals 6 is a half sib of 4 and 5

34
Which individuals to sequence?
Let total number of individuals in population be n, number of individuals that can be sequenced be m. A = average relationship matrix among n individuals, from pedigree c is a vector of size n, which for each animal has the average relationship to the population (eg. Sum up the elements of A down the column for individual i, take mean)
")
35
Which individuals to sequence?
If we choose a group of m animals for sequencing, how much of the diversity do they capture pm = Am-1cm Where Am is the sub matrix of A for the m individuals, and cm is the elements of the c vector for the m individuals Proportion of diversity = pm’1n

36
Which individuals to sequence?
Example

37
Which individuals to sequence?
Then choose set of individuals to sequence (m) which maximise pm’1n Step wise regression Find single individual with largest pi, set ci to zero, next largest pi, set ci to zero….. Genetic algorithm
 which maximise pm’1n. Step wise regression. Find single individual with largest pi, set ci to zero, next largest pi, set ci to zero….. Genetic algorithm.")
38
Which individuals to sequence?
Then choose set of individuals to sequence (m) which maximise pm’1n Step wise regression Find single individual with largest pi, set ci to zero, next largest pi, set ci to zero….. Genetic algorithm No A? Use G
 which maximise pm’1n. Step wise regression. Find single individual with largest pi, set ci to zero, next largest pi, set ci to zero….. Genetic algorithm. No A Use G.")
39
Which individuals to sequence?
Poll Dorset sheep

40
Imputation of full sequence data
Two groups of individuals Sequenced individuals: reference population Individuals genotyped on SNP array: target individuals

41
Imputation of full sequence data
Steps: Step 1. Find polymorphisms in sequence data Step 2. Genotype all sequenced animals for polymorphisms (SNP, Indels) Step 3. Phase genotypes (eg Beagle) in sequenced individuals, create reference file Step 4. Impute all polymorphisms into individuals genotyped with SNP array
 Step 3. Phase genotypes (eg Beagle) in sequenced individuals, create reference file. Step 4. Impute all polymorphisms into individuals genotyped with SNP array.")
42
Imputation of full sequence data
Variant calling SamTools mPileup Vcf file -> filter (number forward /reverse reads of each allele, read depth, quality, filter number of variants in 5bp window) Create BAM files 1. Filter reads on quality score, trim ends 2. Remove PCR duplicates 3. Align with BWA Beagle Phasing in Reference Input genotype probs from Phred scores QC with 800K BAM Reference file for imputation Analysis Genome wide association Genomic selection Beagle Imputation in Target SNP array data in target population Genotype probabilities
 Create BAM files. 1. Filter reads on quality score, trim ends. 2. Remove PCR duplicates. 3. Align with BWA. Beagle Phasing in Reference. Input genotype probs from Phred scores. QC with 800K. BAM. Reference file for imputation. Analysis. Genome wide association. Genomic selection. Beagle Imputation in Target. SNP array data in target population. Genotype probabilities.")
43
Imputation of full sequence data
How accurate?

44
Run4.0 1000 bull genomes Run 4.0 1147 animals sequenced 27 breeds
Breed/Cross Number Holstein (Black and White) 288 Simmental (Dual and Beef) 216 Angus (Black and Red) 138 Jersey 61 Brown Swiss 59 Gelbvieh 34 Charolais 33 Hereford 31 Limousin Guelph Composite 30 Beef Booster 29 Alberta Composite 28 Montbeliarde AyrshireFinnish 25 Normande 24 Holstein (Red and White) 23 Swedish Red 16 Danish Red 15 Other Crosses 11 Belgian Blue 10 Piedmontese 5 Eringer 2 Galloway Unknown Scottish Highland Pezzata Rossa Italiana 1 Romagnola Salers Tyrolean Grey Total 1147 1147 animals sequenced 27 breeds 20 Partners Average 11X CRV
 288. Simmental (Dual and Beef) 216. Angus (Black and Red) 138. Jersey. 61. Brown Swiss. 59. Gelbvieh. 34. Charolais. 33. Hereford. 31. Limousin. Guelph Composite. 30. Beef Booster. 29. Alberta Composite. 28. Montbeliarde. AyrshireFinnish. 25. Normande. 24. Holstein (Red and White) 23. Swedish Red. 16. Danish Red. 15. Other Crosses. 11. Belgian Blue. 10. Piedmontese. 5. Eringer. 2. Galloway. Unknown. Scottish Highland. Pezzata Rossa Italiana. 1. Romagnola. Salers. Tyrolean Grey. Total animals sequenced. 27 breeds. 20 Partners. Average 11X. CRV.")
45
1000 bull genomes Run 4.0 36.9 million filtered variants
35.2 million SNP 1.7 million INDEL X

46
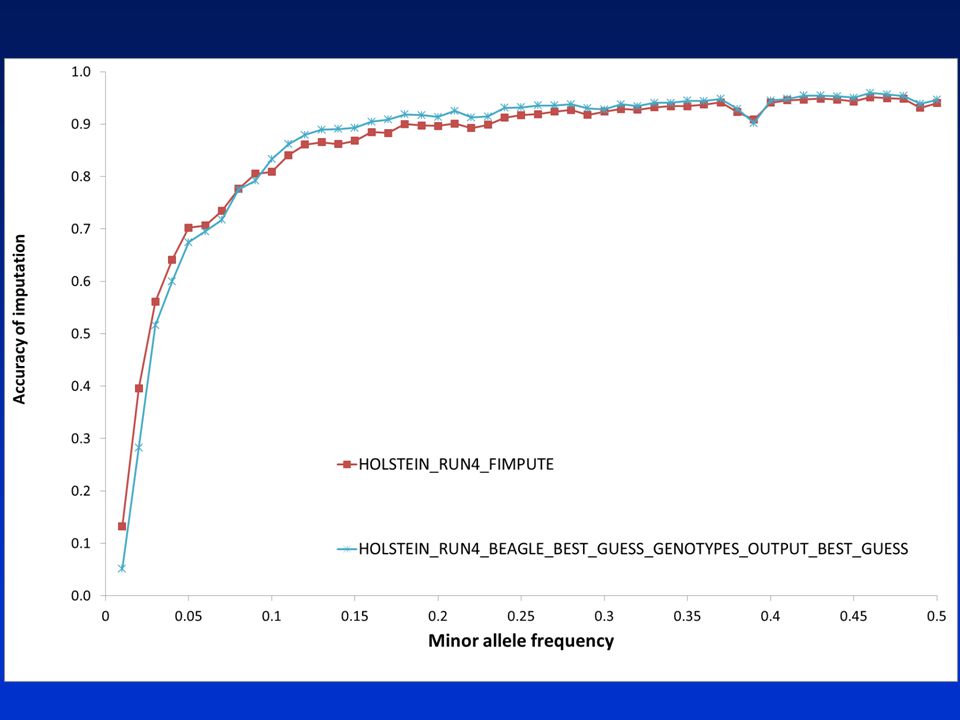
Imputation of full sequence data
Accuracy? Chromosome 14 Remove 50 Holsteins, 20 Jerseys from data set Reduce genotypes to 800K for these animals Impute full sequence using rest of animals as reference 46

47
Imputation of full sequence data
47

48
Imputation of full sequence data
48

49
Imputation of full sequence data
49

50
Imputation of full sequence data
Why so difficult to impute rare mutations? Examples Complex Veterbral Malformation (CVM) and Bovine Leukocyte Deficiency (BLAD) All cases of CVM trace back to Ivanhoe Bell BLAD traces to Osbornedale Ivanhoe 50
 and Bovine Leukocyte Deficiency (BLAD) All cases of CVM trace back to Ivanhoe Bell. BLAD traces to Osbornedale Ivanhoe. 50.")
51
Imputation of full sequence data
Why so difficult to impute rare mutations? BLAD CVM Location Chr1: Chr3: Frequency 0.0014 0.0103 Bulls genotyped 5987 Imputed correctly 5970 5836 Accuracy 0.9972 0.9748 # Carriers 17 123 # Carriers correctly imputed 13 5 Prop. Carriers correctly imputed 0.765 0.041 51

52
Imputation of full sequence data
Why so difficult to impute rare mutations? The BLAD mutation is in a unique 250kb haplotype, which does not occur in any non- carriers The CVM mutation is in a 250kb haplotype which occurs in many non carriers, and also occurs in breeds without mutation Hypothesis – BLAD mutation occurred on rare haplotype, while CVM a recent mutation that occurred on a common haplotype background 52

53
Imputation of full sequence data
Computationally efficient strategies Beagle – run imputation in chromosome segments, say 5MB with 0.5MB overlap (to avoid edge effects) Fimpute – much faster than Beagle, used to impute 32,500 animals from 800K to 16 million SNP! Does not give probabilties Beagle phasing + Minimac 53
 Fimpute – much faster than Beagle, used to impute 32,500 animals from 800K to 16 million SNP! Does not give probabilties. Beagle phasing + Minimac. 53.")
56
Conclusion Impute to fill in missing genotypes low density to high density to save $$ Accuracy depends on size of reference, effective population size, relationship to reference, marker density Imputation to sequence possible, relatively low accuracies for rare alleles Use genotype probabilities from imputation in GWAS and genomic prediction

Similar presentations













 association? How do we test for association? When to use association.>")

>")




