Download presentation
Presentation is loading. Please wait.

2
Determine if a new agent or a new treatment regimen appears sufficiently efficacious to be worth further investigation ◦ Not attempting to prove or establish that the new agent improves outcome Verify the safety of the therapy Provide statistical rigor/formal evaluation context and targeted patient population

3
Often formulate as testing a null hypothesis vs. an alternative ◦ E.g. H 0 : p r = 0.05 vs. H a : p r = 0.20, where p r is the true proportion of patients who will respond to the new agent Consequence of a type I error ( ): an ineffective agent will be studied further ◦ Use = 0.10 (one-sided) ◦ Larger than in phase III studies
: an ineffective agent will be studied further ◦ Use = 0.10 (one-sided) ◦ Larger than in phase III studies.")
4
Consequence of a type II error ( ): an effective agent will not be studied further ◦ should be < 0.10 In practice, tend to be multiple phase II studies performed in multiple diseases, so the overall chance of missing an effective treatment is lower Selection of therapies for phase III testing is based on all available data, not on a single phase II study
: an effective agent will not be studied further ◦ should be < 0.10 In practice, tend to be multiple phase II studies performed in multiple diseases, so the overall chance of missing an effective treatment is lower Selection of therapies for phase III testing is based on all available data, not on a single phase II study")
5
Single arm with single analysis (can have multiple single arm studies in one protocol) Single arm with interim stopping rules (usually with suspension of accrual) Randomized selection designs (pick-the-winner) Comparative randomized control Randomized discontinuation designs
 Single arm with interim stopping rules (usually with suspension of accrual) Randomized selection designs (pick-the-winner) Comparative randomized control Randomized discontinuation designs")
6
Patients refractory to standard therapy If some patients improve, agent must have some activity Often use H 0 : p r = 0.05 vs. H a : p r = 0.20 Simon’s (1989) optimal two-stage designs minimize expected sample size under H 0
 optimal two-stage designs minimize expected sample size under H 0.")
7
Simon’s optimal design for p r = 0.05 vs 0.20: ◦ 1 st stage: treat 12 patients; stop if no responses ◦ 2 nd stage: treat 25 patients; conclude inactive if < 4 / 37 (11%) respond CTEP / IDB has been pushing this design for new agents in diseases without prior evidence of activity
 respond CTEP / IDB has been pushing this design for new agents in diseases without prior evidence of activity")
8
Single arm two-stage designs are inefficient for multicenter studies ◦ Time and effort needed to develop protocol and CRFs and set up database ◦ Cost of activation at institutions Prefer settings where single stage designs are appropriate or studies with multiple strata and / or multiple arms

9
Might be appropriate ◦ If some prior evidence of activity ◦ For combinations of new drugs with standard treatments Example: H 0 : p r = 0.20 vs H a : p r = 0.37 (null rate depends on level of activity for standard rx) ◦ 1-stage: 45 patients, reject H 0 if > 12 / 45 (27%) respond ◦ 2-stage: conclude inactive if < 5 / 25 (20% 1 st stage) or 13/50 (26% overall) respond
 ◦ 1-stage: 45 patients, reject H 0 if > 12 / 45 (27%) respond ◦ 2-stage: conclude inactive if < 5 / 25 (20% 1 st stage) or 13/50 (26% overall) respond")
10
Cytostatic agents might improve disease stabilization rates rather than improve response rates Test for improvement in disease stabilization rates; e.g. H 0 : p s = 0.30 vs. H a : p s = 0.50, where p s = proportion stable or responding (free of progression) at x months (e.g. x = 4) Calculations the same as for response
 at x months (e.g. x = 4) Calculations the same as for response.")
11
Multinomial: test e.g. H 0 : p r = 0.05 and p s = 0.30 vs. H a : p r > 0.05 or p s > 0.30 ◦ Less efficient than binomial ◦ May be more difficult to interpret TTP or PFS ◦ Kaplan-Meier estimate at single time or other nonparametric test ◦ Parametric (e.g. exponential) models can be slightly more efficient Survival generally not appropriate
 models can be slightly more efficient Survival generally not appropriate.")
12
Test e.g. H 0 : p r = 0.05 and p s = 0.30 vs. H a : p r > 0.05 or p s > 0.30 Need to consider power against multiple alternative values; e.g. H a1 : p r = 0.20, p s = 0.30 H a2 : p r = 0.14, p s = 0.40 H a3 : p r = 0.05, p s = 0.50 1-stage: n=46, reject H 0 if > 6 responses or >20 cases responding or stable ◦ = 0.09; power = 0.92 for H a1, H a2, & H a3

13
Separate evaluation of each arm ◦ Each arm evaluated in a similar population Selection designs: select the ‘best’ arm for further study Comparative randomized control Randomized discontinuation Randomized designs are larger and more complex – need to explain each arm to patients

14
Concern about selection bias in studies without a simultaneous control group ◦ Studies can enroll different patient groups even with the same nominal population ◦ Population drift and stage migration Control groups more appropriate for evaluating contribution to a combination or effect on progression than for determining if any response activity Comparing studies from different groups

15
Often not needed because ◦ Phase II studies can only detect fairly large effects, so biases would need to be large ◦ Consequence of a false positive is further testing of an inactive drug ◦ Cooperative group or other studies conducted in the same network with central data review produce fairly consistent results Increase the time and expense for phase II evaluation

16
(Simon, Wittes and Ellenberg, 1985) randomize between 2 or more experimental arms (no control arm) ◦ In a sense, least efficacious arm is a control for the others Select the best arm for further evaluation Usually define ‘best’ to be the arm with the best outcome, no matter how small the difference
 randomize between 2 or more experimental arms (no control arm) ◦ In a sense, least efficacious arm is a control for the others Select the best arm for further evaluation Usually define ‘best’ to be the arm with the best outcome, no matter how small the difference")
17
With two arms, 0.50 ◦ Rationale: doesn’t matter which arm is selected if they are nearly equivalent Often separate efficacy test for each arm, too ◦ 1-stage or 2-stage Usually prefer randomizing over a series of separate studies ◦ Facilitates (informal) comparisons ◦ Guards against sampling bias
 comparisons ◦ Guards against sampling bias")
18
RANDOMIZERANDOMIZE RX1 RX2 RXk... Estimated Resp Rate R1/n1 R2/n2 Rk/nk... RXj is ‘best’ if Rj/nj > Ri/ni for i j Can use other endpoints

19
Example: Simon’s optimal 2-stage design for H 0 : p r = 0.20 vs H a : p r = 0.40 enrolls 17 patients in the 1 st stage and 20 in the 2 nd ( = =.10) Apply this design to each arm in a 2-arm randomized selection design Prob arm is winner p r1 p r2 RX1RX2Neither.20.40.015.890.095.30.40.147.758.095
 Apply this design to each arm in a 2-arm randomized selection design Prob arm is winner p r1 p r2 RX1RX2Neither")
20
Probability of selecting the best arm declines as the number of arms increases P{X 1 >max(X 2,…,X k )} = x P(X 1 =x)P(X 2 <x, X 3 <x…,X k <x|X 1 =x) = x P(X 1 =x)P(X 2 <x) P(X 3 <x)… P(X k <x) = x P(X 1 =x)P(X 2 <x) k-1 if X 2, …, X k have the same distribution
} = x P(X 1 =x)P(X 2 <x, X 3 <x…,X k <x|X 1 =x) = x P(X 1 =x)P(X 2 <x) P(X 3 <x)… P(X k <x) = x P(X 1 =x)P(X 2 <x) k-1 if X 2, …, X k have the same distribution")
21
X 1 ~Bin(50,.32); X 2,…,X k ~Bin(50,.20) gives P{X 1 >max(X 2,…,X k )} =.90 for k = 2 and P{X 1 >max(X 2,…,X k )} =.72 for k = 6 Advanced renal trial of several targeted agents: 6 arms, n=55 / arm ◦ TTP compared via Cox model ◦ If one arm has median TTP of 7.2 months and the other 5 have median TTP of 4.8 months (50% improvement), then the probability of selecting the best arm is 0.87
; X 2,…,X k ~Bin(50,.20) gives P{X 1 >max(X 2,…,X k )} =.90 for k = 2 and P{X 1 >max(X 2,…,X k )} =.72 for k = 6 Advanced renal trial of several targeted agents: 6 arms, n=55 / arm ◦ TTP compared via Cox model ◦ If one arm has median TTP of 7.2 months and the other 5 have median TTP of 4.8 months (50% improvement), then the probability of selecting the best arm is 0.87")
22
Discussed for evaluating cytostatic agents in Korn et al. (2001) Randomize experimental vs. standard and formally compare the arms Appropriate if don’t have a reasonable prior estimate of expected control arm outcomes Endpoint could be any of the standard phase II endpoints (e.g. TTP, response) Might target larger differences than a phase III
 Randomize experimental vs. standard and formally compare the arms Appropriate if don’t have a reasonable prior estimate of expected control arm outcomes Endpoint could be any of the standard phase II endpoints (e.g. TTP, response) Might target larger differences than a phase III.")
23
Test could be a definitive (phase III) evaluation with < 0.025 (one-sided) ◦ If little prior phase II efficacy data, need early stopping rules for lack of benefit ◦ Might not be appropriate if a second phase III study evaluating survival would be needed
 evaluation with < (one-sided) ◦ If little prior phase II efficacy data, need early stopping rules for lack of benefit ◦ Might not be appropriate if a second phase III study evaluating survival would be needed")
24
Test could be a suggestive (phase II) evaluation with a larger (e.g. 0.10 to 0.20) ◦ Appropriate for screening new agents ◦ If positive, still needs to be followed by a definitive phase III study ◦ Korn et al. suggest using = 0.20, because the sample size with = 0.10 is large enough that it might be better to go directly to the definitive study
 ◦ Appropriate for screening new agents ◦ If positive, still needs to be followed by a definitive phase III study ◦ Korn et al. suggest using = 0.20, because the sample size with = 0.10 is large enough that it might be better to go directly to the definitive study.")
25
3-arm comparison of TTP (two dose levels of bevacizumab), targeting a large difference (100% improvement in median TTP), but designed to be definitive (Yang, 2003) ◦ Overall =.05 (two-sided), = 0.20 ◦ Each comparison at one-sided 0.0125 ◦ Needed about 50 patients / arm (stopped early because of highly significant results) ◦ Crossover from placebo to low dose drug
, targeting a large difference (100% improvement in median TTP), but designed to be definitive (Yang, 2003) ◦ Overall =.05 (two-sided), = 0.20 ◦ Each comparison at one-sided ◦ Needed about 50 patients / arm (stopped early because of highly significant results) ◦ Crossover from placebo to low dose drug")
26
Was overall =.05 appropriate? ◦ A second, larger study is still needed for survival ◦ Could have identified drug as promising with even fewer patients (larger )
.")
27
Was a placebo needed? ◦ Evaluation bias should be much smaller than a doubling of TTP ◦ May not be to identify promising drugs ◦ FDA tends to require a placebo for TTP Was the control arm needed? ◦ Would results from a single arm, single institution study have been convincing?

28
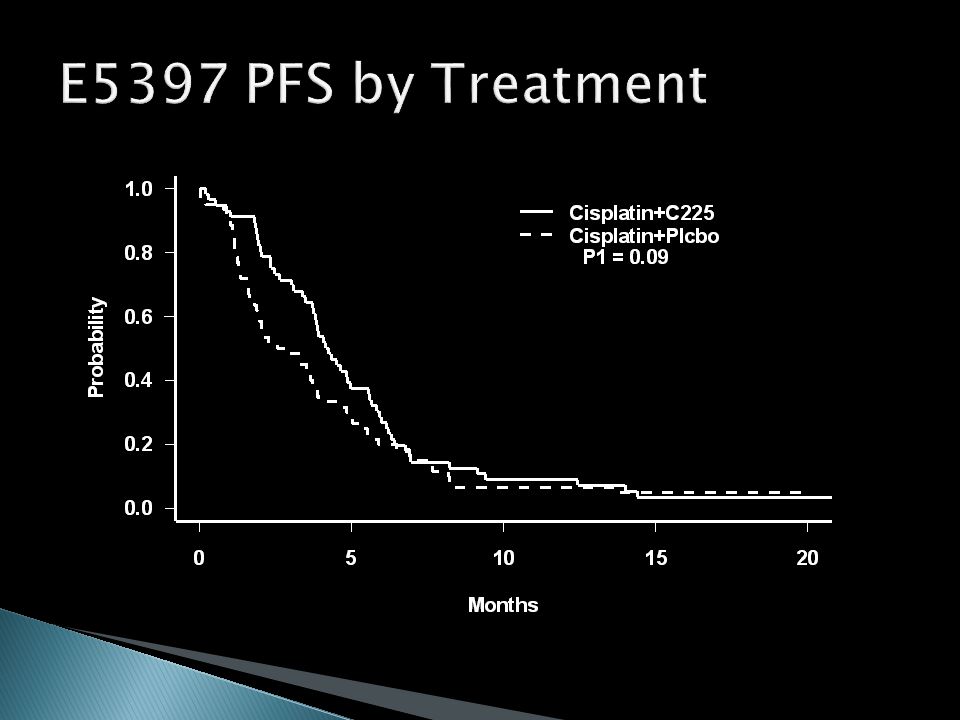
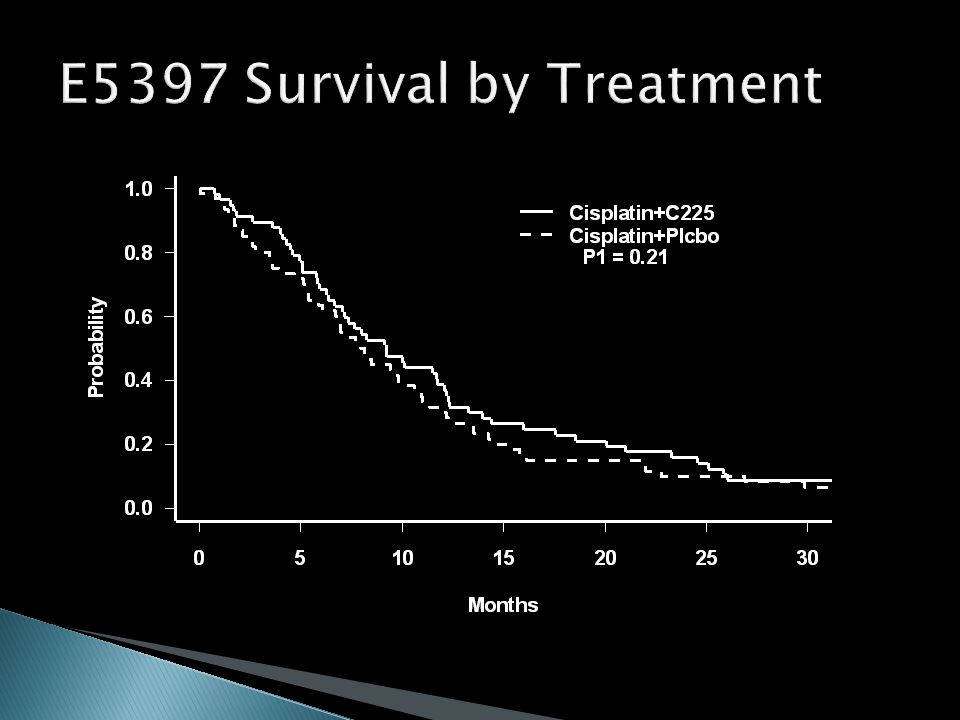
Cisplatin + C225 vs. Cisplatin + Placebo Designed to have 90% power to detect an improvement in median PFS from 2 months to 4 months (100% improvement) with = 0.025 (one-sided) With allowance for non-compliance, required 54 eligible patients / arm Final accrual was 117 eligible patients
 with = (one-sided) With allowance for non-compliance, required 54 eligible patients / arm Final accrual was 117 eligible patients.")
29
Cisplatin + C225 Cisplatin + Placebo P-value (one-sided) Response Rate26%10%0.02 Median PFS4.2 mos2.7 mos0.09 Median Survival9.2 mos8.0 mos0.21 Hazard Ratios (Placebo/C225) and 95% CIs PFS: 1.31 (0.91, 1.89) Survival: 1.16 (0.80, 1.69)
 Response Rate26%10%0.02 Median PFS4.2 mos2.7 mos0.09 Median Survival9.2 mos8.0 mos0.21 Hazard Ratios (Placebo/C225) and 95% CIs PFS: 1.31 (0.91, 1.89) Survival: 1.16 (0.80, 1.69)")
32
Study is not definitive – underpowered for both PFS and survival Is it promising – should a follow-up study of C225 be done? Would a better strategy have been a single arm phase II with a response endpoint, followed by a definitive phase III based on the ‘promising’ response rate of 26%?

33
PFS reaches the one-sided = 0.10 cutoff for a ‘promising’ phase II result Survival would not have been an appropriate endpoint ◦ Estimated improvement is 16% ◦ Confidence interval consistent with 20% decrease to a 69% increase ◦ Phase II sample sizes are not adequate to detect realistic survival effects

34
An enrichment strategy based on randomizing patients who appear to be doing well on the treatment (Rosner, Stadler, Ratain, 2002) Initially all patients are treated, patients free of progression for some period of time are randomized between continuing treatment and placebo, with crossover from placebo to treatment at progression or specified PFI Complex design with a blinded randomization and 3 registration points
 Initially all patients are treated, patients free of progression for some period of time are randomized between continuing treatment and placebo, with crossover from placebo to treatment at progression or specified PFI Complex design with a blinded randomization and 3 registration points")
35
RANDOMIZERANDOMIZE RX Placebo REGISTERREGISTER REASSESREASSES Initial RX Off study PD SD Continue RX Response Crossover at PD or after specified PFI (run-in)
")
36
Usefulness depends on how successful the run-in is in selecting patients benefiting from treatment ◦ TTP is highly variable in most diseases, so randomized population will be a mixture ◦ Korn et al. (2001), Capra (2004) suggest often less efficient than standard RCT Carry-over effect could dilute difference between randomized arms Requires much larger sample size
, Capra (2004) suggest often less efficient than standard RCT Carry-over effect could dilute difference between randomized arms Requires much larger sample size.")
37
CALGB 69901 (CAI in RCC) Randomize patients if stable after 16 weeks Enrolled 374 patients; randomized 65 eligible patients (17%) ◦ Enrichment strategy was not successful, but does CAI have any activity? ◦ Did they learn any more from 374 patients than ECOG did from 57 patients in a more traditional two-stage phase II design (E4896)?
 .")
38
In many settings, conventional phase II designs may still be appropriate Start-up costs for single-arm two-stage designs are a concern Randomized phase II studies allow evaluation of multiple agents or schedules and protect against sampling bias Selection designs are useful for informal comparison and identifying promising agents

39
Control arms should not ordinarily be needed, but can be effective in some settings Survival is seldom (never?) the best phase II endpoint Randomized discontinuation designs may not be appropriate and need to be strongly justified
 the best phase II endpoint Randomized discontinuation designs may not be appropriate and need to be strongly justified")
40
Capra WB (2004). Comparing the power of the discontinuation design to that of the classic randomized design on time-to-event endpoints. Controlled Clinical Trials 25:168-177. Freidlin B, Dancey J, Korn EL, Zee B, Eisenhauer E (2002) Multinomial phase II trial designs (letter to the editor). Journal of Clinical Oncology 20:599. Korn EL, Arbuck SG, Pluda JM, Simon R, Kaplan RS, Christian MC (2001). Clinical trial designs for cytostatic agents: are new approaches needed? Journal of Clinical Oncology 19:265-272. Rosner GL, Stadler W, Ratain MJ (2002). Randomized discontinuation design: application to cytostatic antineoplastic agents. Journal of Clinical Oncology 20:4478-4484. Simon R (1989). Optimal two-stage designs for phase II clinical trials. Controlled Clinical Trials 10:1-10. Simon R, Wittes RE, Ellenberg SS (1985). Randomized phase II clinical trials. Cancer Treatment Reports 12:1375-1381. Yang JC et al. (2003). A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. New England Journal of Medicine 349:427-434.
 Multinomial phase II trial designs (letter to the editor). Journal of Clinical Oncology 20:599. Korn EL, Arbuck SG, Pluda JM, Simon R, Kaplan RS, Christian MC (2001). Clinical trial designs for cytostatic agents: are new approaches needed. Journal of Clinical Oncology 19: Rosner GL, Stadler W, Ratain MJ (2002). Randomized discontinuation design: application to cytostatic antineoplastic agents. Journal of Clinical Oncology 20: Simon R (1989). Optimal two-stage designs for phase II clinical trials. Controlled Clinical Trials 10:1-10. Simon R, Wittes RE, Ellenberg SS (1985). Randomized phase II clinical trials. Cancer Treatment Reports 12: Yang JC et al. (2003). A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. New England Journal of Medicine 349:")
Similar presentations

 regarding unknown population parameter values based on sample data Elements of a hypothesis test: Null hypothesis.>")





 Design. Prospective Co-Development of Drugs and Companion Diagnostics 1. Develop a completely specified genomic classifier of the.>")


:>")









