Download presentation
Presentation is loading. Please wait.

1
Inherited Diseases of Muscle: Histologic Features David Lacomis, MD

2
Classification of Myopathies ACQUIREDINHERITED Inflammatory Myopathies Dystrophies Polymositis (PM) Dystrophinopathies Dermatomyositis (DM) Limb-Girdle Inclusion body myositis (IBM) Myotonic Granulomatous myositis Facioscapulohumeral (FSHD) Infectious myositis Oculopharyngeal (OPD) Toxic Distal EndocrineCongenital Metabolic Mitochondrial Glycogen & lipid storage
 Dystrophinopathies Dermatomyositis (DM) Limb-Girdle Inclusion body myositis (IBM) Myotonic Granulomatous myositis Facioscapulohumeral (FSHD) Infectious myositis Oculopharyngeal (OPD) Toxic Distal EndocrineCongenital Metabolic Mitochondrial Glycogen & lipid storage")
3
Opaque or hyaline fibers Increase in endomysial connective tissue Frozen Section from a Patient with Duchenne Muscular Dystrophy Group of basophilic regenerating fibers

4
Normal Immunohistochemical Stain for Dystrophin Subsarcolemmal staining

5
Duchenne Muscular Dystrophy Absent staining for dystrophin

6
split fiber (non-specific chronic change) Becker Muscular Dystrophy Reduced but present staining
 Becker Muscular Dystrophy Reduced but present staining")
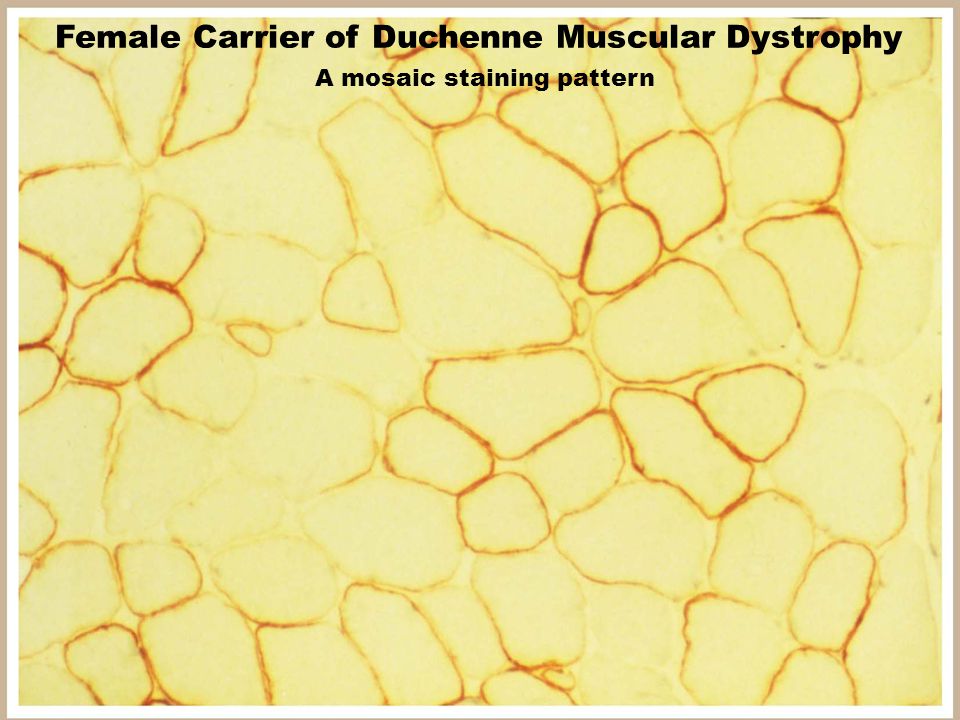
7
Female Carrier of Duchenne Muscular Dystrophy A mosaic staining pattern

9
INHERITANCE GENETIC ABNORMALITY DISORDER X-linkedDystrophin Emerin Duchenne, Becker MD Emery-Dreifuss MD ADMyotilin Lamin A/C Caveolin – 3 PABP2 -crystallin/Desmin Limb-Girdle MD (LGMD 1A) LGMD 1B LGMD 1C Oculopharyngeal Myofibrillar Myopathy ARCalpain – 3 Dysferlin Sarcoglycan a Sarcoglycan Sarcoglycan Δ Sarcoglycan Telethonin Fukuitin-rel prot LGMD 2A LGMD 2B LGMD 2C LGMD 2D LGMD 2E LGMD 2F LGMD 2G LGMD 2H LGMD 2I Mutations in “Limb-Girdle” & Other Dystrophies
 LGMD 1B LGMD 1C Oculopharyngeal Myofibrillar Myopathy ARCalpain – 3 Dysferlin Sarcoglycan a Sarcoglycan Sarcoglycan Δ Sarcoglycan Telethonin Fukuitin-rel prot LGMD 2A LGMD 2B LGMD 2C LGMD 2D LGMD 2E LGMD 2F LGMD 2G LGMD 2H LGMD 2I Mutations in Limb-Girdle & Other Dystrophies")
10
Sarcolemma nucleus Lamin A/C (emerin) sarcoglycans Dystroglycan complex Laminin-2 Extracellular Matrix Dysferlin Caveolin 3 Actin Dystrophin Locations of Affected Proteins in Muscular Dystrophies
 sarcoglycans Dystroglycan complex Laminin-2 Extracellular Matrix Dysferlin Caveolin 3 Actin Dystrophin Locations of Affected Proteins in Muscular Dystrophies")
11
Emery-Dreifuss Muscular Dystrophy Gomori trichrome-stained frozen section Necrotic fiber Variation in fiber size with many hypertrophic fibers Increase in endomysial connective tissue Nonspecific so-called dystrophic changes seen in many of the muscular dystrophies. Can also be seen in any chronic myopathic disorder. This disorder is due to loss of the protein emerin.

12
Myotonic Dystrophy Chronic changes Marked excess in internalized nuclei Variation in fiber sizes Nuclear clumps (not shown)
")
13
H & E, paraffin The excess of internalized nuclei can lead to nuclear chains.

14
Myotonic Dystrophy NADH-reacted section Ring fibers in which myofilaments are organized in different directions

15
Fascioscapulohumeral Dystrophy (FSHD) The majority of dystrophies do not have a specific histopathologic appearance. Clinical features are also very important. For example, winging of the scapula is characteristic of FSHD.

16
FSH Dystrophy Variable non-specific changes Range from scattered atrophy to “dystrophic” features. Inflammation can be present.

17
Basophilic subsarcolemmal structures are sarcoplasmic masses. Sometimes occur in chronic myopathies such as FSH and myotonic dystrophy.

18
Sarcoplasmic Masses Stained darkly with NADH reaction

19
Oculopharyngeal Muscular Dystrophy (OPD) Variation in muscle fiber size with atrophic angulated fibers Sometimes contain rimmed vacuoles
 Variation in muscle fiber size with atrophic angulated fibers Sometimes contain rimmed vacuoles")
20
Higher power view of Gomori trichrome-stained section Angulated fibers Fiber containing a large rimmed vacuole

21
Oculopharyngeal Dystrophy Gomori trichrome Ragged red fibers are sometimes seen. Characteristic of proliferation of abnormal mitochondria.

22
May be identified by electron microscopy in OPD Intranuclear Filamentous Inclusions

23
Central areas of absent staining in the dark type I fibers Mitochondria absent Congenital Myopathies: Central Core Myopathy NADH

24
The core consists of disorganized myofibrils and the area is devoid of mitochondria.

25
Congenital Fiber Type Disproportion H&E Bimodal size population

26
Smaller fibers are type I More numerous Stain lightly Larger or normal fibers are type II Congenital Fiber Type Disproportion ATPase pH 4.3

27
Eosinophilic inclusions present Nemaline Myopathy

28
Eosinophilic inclusions stain darkly Nemaline Myopathy Gomori trichrome

29
Named for thread-like appearance Inclusions extend from Z-band to Z-band Nemaline Myopathy Electron microscopy

30
Muscle Biopsy from an Infant Internalized nuclei predominant Consistent with centronuclear myopathy Can be seen in other disorders such as myotonic dystrophy with congenital onset

32
Muscle Biopsy from an Infant: Centronuclear Myopathy Central position of the nucleus resembling an embryonic myotube

33
Metabolic: Inherited – Mitochondrial MELAS Syndrome Ragged red fiber present

34
SDH-rich fibers are seen with mitochondrial proliferation MELAS Syndrome Succinic dehydrogenase reaction

36
“Ragged-red” Fibers H&E

37
SDH-rich Fibers

38
Cox Normal Fibers

39
Many COX-negative Fibers COX-negative fibers are usually seen with mtDNA mutations.

40
Aggregates of mitochondria containing paracrystalline inclusions are frequent. Non-specific Mitochondrial Disorders Electron Microscopy

41
Higher power view of paracrystalline inclusion

42
Increased lipid storage Seen in carnitine deficiency states (primary or secondary) Sometimes as a consequence of certain toxins Focal increases can be non-specific Oil-red-O stain
 Sometimes as a consequence of certain toxins Focal increases can be non-specific Oil-red-O stain")
43
Lipid Storage Myopathy Electron microscopy

44
Some glycogen storage myopathies, such as myophosphorylase deficiency (McArdle’s Disease), cause subsarcolemmal blebs. PAS-positive due to the presence of glycogen. Glycogen Storage Myopathies

45
McArdles Disease: Phosphorylase Reaction Normal Control Disease (Absent)
")
46
Subsarcolemmal collection of glycogen is shown. McArdle’s Disease Electron Microscopy

47
Acid Maltase Deficiency Acid phosphatase Due to the intralysosomal activity of this enzyme Prominent staining with acid phosphatase in vacuoles Vacuolar myopathy noted.

48
Normal Glycogen PAS stain (control)
")
49
Increased Glycogen Acid maltase deficiency Increased glycogen (diffusely and in vacuoles)
")
50
Glycogen is digested by diastase in most glycogen storage diseases.

51
Aggregates of Glycogen within Autophagic Vacuoles (Acid Maltase Deficiency) Electron microscopy
 Electron microscopy")
52
Tubular aggregates occur in an inherited myopathy, non- specifically, and in some patients with myalgias. Miscellaneous Disorder

53
Bright red with Gomori trichrome

54
Stain darkly with NADH, no staining with SDH Miscellaneous Disorder

55
Tubular Aggregates Via Electron Microscopy

Similar presentations






 Wenya Hou Xue Jing Yitang Wang Jiezhong Zhang.>")


 Unlike: neuronopathies: secondary.>")











