Download presentation
Presentation is loading. Please wait.

1
Blood Drugs

2
describes drugs that are useful in treating three important dysfunctions of blood: thrombosis, bleeding, and anemia.

3
Drugs used in treatment of thrombosis

4
Objectives To learn how Blood Clots are formed and broken down ?
What drugs can be used to regulate clotting ?

5
THROMBOSIS Thrombosis is the formation of an unwanted clot within a blood vessel. it is the most common abnormality of hemostasis.

6
Consequences of thrombus
angina Myocardial infarction stroke Deep venous thrombosis

7
Thrombus versus embolus
A clot that adheres to a vessel wall is called a thrombus whereas an intravascular clot that floats in the blood is termed an embolus. Thus, a detached thrombus becomes an embolus. Both thrombi and emboli are dangerous, because they may occlude blood vessels and deprive tissues of oxygen and nutrients.

8
Blood Clotting Vascular Phase Platelet Phase Coagulation Phase Fibrinolytic Phase

9
Vascular Phase Vasoconstriction
Exposure of tissues activate Tissue factor and initiate coagulation Tissue Factor

10
Platelet activation 1- resting platelets:
In the absence of injury, resting platelets circulate freely, because the balance of chemical signals indicates that the vascular system is not damaged.

11
A- Chemical mediators synthesized by endothelial cells:
Chemical mediators, such as prostacyclin and nitric oxide, are synthesized by intact endothelial cells and act as inhibitors of platelet aggregation. Prostacyclin (prostaglandin I2) acts by binding to platelet membrane receptors that are coupled to the synthesis of cyclic adenosine monophosphate (cAMP). Elevated levels of intracellular cAMP are associated with a decrease in intracellular Ca2+. This leads to inhibition of platelet activation and inhibit release of platelet aggregation agents.
 acts by binding to platelet membrane receptors that are coupled to the synthesis of cyclic adenosine monophosphate (cAMP). Elevated levels of intracellular cAMP are associated with a decrease in intracellular Ca2+. This leads to inhibition of platelet activation and inhibit release of platelet aggregation agents.")
12
Damaged endothelial cells synthesize less prostacyclin, resulting in a localized reduction in prostacyclin levels. The binding of prostacyclin to platelet receptors is decreased, resulting in lower levels of intracellular cAMP, which leads to platelet aggregation.

13
B- Roles of thrombin, thromboxane, and collagen:
The platelet membrane also contains receptors that can bind thrombin, thromboxane2, and exposed collagen, that when occupied triggers platelet aggregation. In the intact, normal vessel, circulating levels of thrombin and thromboxane are low the intact endothelium covers the collagen in the subendothelial layers. The corresponding platelet receptors are thus unoccupied and remain inactive; as a result, platelet activation and aggregation are not initiated.

14
2- Platelet adhesion When the endothelium is injured, platelets adhere to and virtually cover the exposed collagen of the subendothelium. This triggers a complex series of chemical reactions, resulting in platelet activation

15
3- platelet activation Receptors on the surface of the adhering platelets are activated by the collagen of the underlying connective tissue. This causes morphologic changes in the platelets and the release of platelet granules containing chemical mediators, such as: adenosine diphosphate (ADP) thromboxane A2 Serotonin platelet-activation factor thrombin. .
 thromboxane A2. Serotonin. platelet-activation factor. thrombin. .")
16
These signaling molecules bind to receptors in the outer membrane of resting platelets circulating nearby. The previously dormant platelets become activated and start to aggregate

17
4- Platelet aggregation
Platelet activation leads to : activation of the glycoprotein (GP) IIb/IIIa receptors that bind fibrinogen and, ultimately, regulate platelet-platelet interaction and thrombus formation. Fibrinogen, simultaneously binds to GP IIb/IIIa receptors on two separate platelets, resulting in platelet cross-linking and platelet aggregation. This leads to an avalanche of platelet aggregation, because each activated platelet can recruit other platelets
 IIb/IIIa receptors that bind fibrinogen and, ultimately, regulate platelet-platelet interaction and thrombus formation. Fibrinogen, simultaneously binds to GP IIb/IIIa receptors on two separate platelets, resulting in platelet cross-linking and platelet aggregation. This leads to an avalanche of platelet aggregation, because each activated platelet can recruit other platelets.")
18
Role of Platelet Activation and Aggregation in Ischemic Syndromes
Figure 1. Role of Platelet Activation and Aggregation in Ischemic Syndromes. Platelets flow in the blood as smooth disks until they are activated and change their conformation into spiny spheres (Panel A). Healthy endothelium secretes nitric oxide (NO) and prostacyclin (PGI2), which help keep the platelets in an inactive state. CD39 on the endothelial surface converts adenosine diphosphate (ADP) a powerful activator of platelets into adenosine monophosphate (AMP); CD73 converts AMP into adenosine, which further prevents the activation of platelets. Injury to the endothelium exposes numerous ligands such as von Willebrand factor and collagen, which can bind their respective receptors, such as glycoprotein Ib/IX/V and glycoprotein VI, on platelets (Panel B). The activated platelet then releases numerous prothrombotic factors, such as thromboxane A2 (TxA2) and ADP, which themselves further activate platelets and amplify the thrombotic process (Panel C). Cross-linking by means of fibrinogen binding to the glycoprotein IIb/IIIa receptor on platelets leads to the formation of platelet aggregates (Panel D). The P2Y12 subtype of the ADP receptor is the site of action of both prasugrel and clopidogrel, as well as of other compounds currently in development (Panel E). Both prasugrel and clopidogrel are thienopyridines that are prodrugs (Panel F). Their active metabolites bind irreversibly to the P2Y12 ADP receptor and inhibit platelet activation and subsequent aggregation. Bhatt D. N Engl J Med 2007;357:
. Healthy endothelium secretes nitric oxide (NO) and prostacyclin (PGI2), which help keep the platelets in an inactive state. CD39 on the endothelial surface converts adenosine diphosphate (ADP) a powerful activator of platelets into adenosine monophosphate (AMP); CD73 converts AMP into adenosine, which further prevents the activation of platelets. Injury to the endothelium exposes numerous ligands such as von Willebrand factor and collagen, which can bind their respective receptors, such as glycoprotein Ib/IX/V and glycoprotein VI, on platelets (Panel B). The activated platelet then releases numerous prothrombotic factors, such as thromboxane A2 (TxA2) and ADP, which themselves further activate platelets and amplify the thrombotic process (Panel C). Cross-linking by means of fibrinogen binding to the glycoprotein IIb/IIIa receptor on platelets leads to the formation of platelet aggregates (Panel D). The P2Y12 subtype of the ADP receptor is the site of action of both prasugrel and clopidogrel, as well as of other compounds currently in development (Panel E). Both prasugrel and clopidogrel are thienopyridines that are prodrugs (Panel F). Their active metabolites bind irreversibly to the P2Y12 ADP receptor and inhibit platelet activation and subsequent aggregation. Bhatt D. N Engl J Med 2007;357:")
21
ANTIPLATELET THERAPY

22
Platelet aggregation inhibitors
Platelet aggregation inhibitors decrease the formation or the action of chemical signals that promote platelet aggregation. The most important of these is the GP IIb/IIIa receptor that ultimately regulates platelet-platelet interaction and thrombus formation.

23
The platelet aggregation inhibitors described below act by:
Inhibit cyclooxygenase-1 (COX-1) or block GP IIb/IIIa or block ADP receptors Thereby interfering in the signals that promote platelet aggregation. Since these agents have different mechanisms of actions, synergistic or additive effects may be achieved when agents from different classes are combined.
 or. block GP IIb/IIIa or. block ADP receptors. Thereby interfering in the signals that promote platelet aggregation. Since these agents have different mechanisms of actions, synergistic or additive effects may be achieved when agents from different classes are combined.")
24
These agents are beneficial in
the prevention and treatment of occlusive cardiovascular diseases in the maintenance of vascular grafts and arterial patency As adjuncts to thrombin inhibitors or thrombolytic therapy in myocardial infarction.

25
A- aspirin Activation of platelets results in stimulation of platelet membrane phospholipases that liberate arachidonic acid from membrane phospholipids. Arachidonic acid is converted into thromboxane A2 by COX enzyme Activated platelets releases thromboxane which activates further platelets. Aspirin inhibits thromboxane A2 synthesis by irreversible inhibition of platelet COX enzyme. This shifts the balance of chemical mediators to favor the antiaggregatory effects of prostacyclin, thus impeding platelet aggregation. \

28
The aspirin-induced suppression of thromboxane last for the life of the anucleated platelet; approximately 7 to 10 days. Aspirin is currently employed in the prophylactic treatment of : transient cerebral ischemia to reduce the incidence of recurrent myocardial infarction and to decrease mortality in pre and post myocardial infarct patients. The recommended dose of aspirin ranges from 81 to 325 mg, with side effects determining the dose chosen.

29
Side effects: Bleeding time is prolonged by aspirin treatment, causing complications that include increased incidence of hemorrhagic stroke as well as gastrointestinal bleeding, especially at higher doses of the drug. NSAID, such as ibuprofen, inhibit COX-1 by transiently competing at the catalytic site. Ibuprofen, if taken concomitantly with, or 2 hours prior to aspirin, can obstruct the access of aspirin to the catalytic site and, thereby, antagonize the platelet inhibition by aspirin.

30
Therefore, aspirin should be taken at least 30 minutes before ibuprofen or at least 8 hours after ibuprofen. Although celecoxib (a selective COX-2 inhibitor) does not interfere in the antiaggregation activity of aspirin there is some evidence that it may contribute to cardiovascular events by shifting the balance of chemical mediators in favor of thromboxane A2.
 does not interfere in the antiaggregation activity of aspirin there is some evidence that it may contribute to cardiovascular events by shifting the balance of chemical mediators in favor of thromboxane A2.")
31
Aspirin: Primary prevention of MI in high risk persons
Secondary prevention of MI, & stroke Clopidogrel: for persons who can’t take aspirin Aspirin+clopidogrel: Acute coronary syndromes

32
Treatment failures occur with aspirin therapy; approximatly 40% of human patients on aspirin therapy develop an ischemic event. There are many possible causes for this. Aspirin is a relatively weak inhibitor of platelet function. It inhibits only one pathway of platelet activation and aggregation. Moreover, platelet aggregation is only one pathway of thrombus formation.

33
True „aspirin resistance‟ refers to failure of aspirin to inhibit TXA2 production.
Potential mechanisms include: (1) decreased bioavailability (2) competition with other NSAIDs (3) accelerated platelet turnover, introducing newly-formed, non aspirinated platelets into the circulation (4) TXA2 production by the aspirin-insensitive COX-2 in newly-formed platelets.
 decreased bioavailability. (2) competition with other NSAIDs. (3) accelerated platelet turnover, introducing newly-formed, non aspirinated platelets into the circulation. (4) TXA2 production by the aspirin-insensitive COX-2 in newly-formed platelets.")
34
Ticlopidine and clopidogrel
Mechanism of action: These drugs irreversibly inhibit the binding of ADP to its receptors on platelets Therapeutic use: Ticlopidine is approved for the prevention of transient ischemic attacks and strokes for patients with prior cerebral thrombotic event. It is also used as adjunct therapy with aspirin following coronary stent implantation to decrease the incidence of stent thrombosis.

36
However, due to its life-threatening hematologic adverse reactions, including neutropenia/agranulocytosis, thrombotic thrombocytopenic purpura (TTP), and aplastic anemia, it is generally reserved for patients who are intolerant to other therapies
, and aplastic anemia, it is generally reserved for patients who are intolerant to other therapies")
37
Clopidogrel is approved for prevention of atherosclerotic events following recent myocardial infarction, stroke, or established peripheral arterial disease. It is also approved for prophylaxis of thrombotic events in acute coronary syndrome (unstable angina). Additionally, clopidogrel is used to prevent thrombotic events associated with percutaneous coronary intervention with or without coronary stent.

38
Compared to ticlopidine, clopidogrel is the preferred agent in ischemic heart disease events, because there is more data to support use of clopidogrel in these cardiac patients. Furthermore, clopidogrel has a better overall side-effect profile although TTP may also occur with this agent.

39
Pharmacokinetics: Food interferes with the absorption of ticlopidine but not with clopidogrel. After oral ingestion, both drugs are extensively bound to plasma proteins. They undergo hepatic metabolism by the cytochrome P450 system to active metabolites that are yet to be identified. The maximum effect is achieved in 3 to 5 days when treatment is suspended, the platelet system requires time to recover.

40
Side effects Ticlopidine has a black box warning due to the severe hematologic adverse reactions associated with its use. Both drugs can cause prolonged bleeding for which there is no antidote. Serious adverse effects of ticlopidine include neutropenia, TTP, and aplastic anemia requiring frequent blood monitoring, especially during the first 3 months of treatment.

41
Drug interaction: Because these drugs can inhibit cytochrome P450, they may interfere with the metabolism of drugs such as phenytoin, tolbutamide, warfarin, fluvastatin, and tamoxifen if taken concomitantly. Indeed, phenytoin toxicity has been reported when taken with ticlopidine.

42
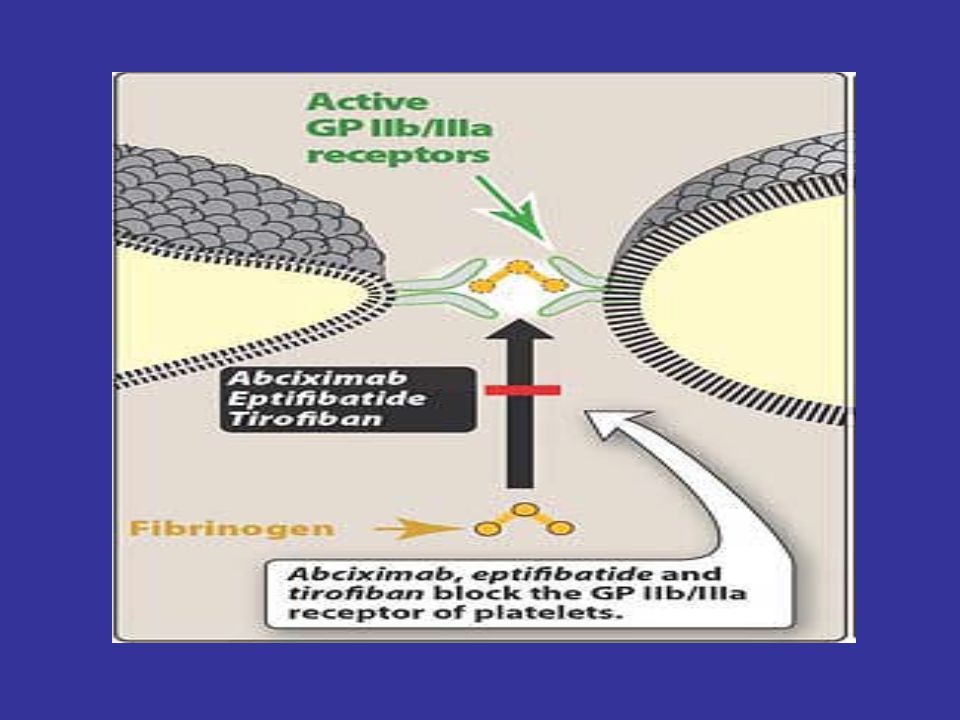
Abciximab chimeric monoclonal antibody
composed of the constant regions of human immunoglobulin joined to the Fab fragments of a murine monoclonal antibody directed against the GP IIb/IIIa complex. By binding to GP IIb/IIIa, the antibody blocks the binding of fibrinogen; consequently, aggregation does not occur

43
Abciximab is given intravenously along with heparin or aspirin as an adjunct to percutaneous coronary intervention for the prevention of cardiac ischemic complications. After cessation of infusion, platelet function gradually returns to normal, with the antiplatelet effect persisting for 24 to 48 hours. The major adverse effect of abciximab therapy is the potential for bleeding, especially if the drug is used with anticoagulants or if the patient has a clinical hemorrhagic condition. Abciximab is expensive, limiting its use in some settings.

44
Eptifibatide and tirofiban
These two antiplatelet drugs act similarly to abciximab, blocking the GP IIb/IIIa receptor. These compounds, like abciximab, can decrease the incidence of thrombotic complications associated with acute coronary syndromes. When intravenous infusion is stopped, these agents are rapidly cleared from the plasma, but their effect can persist for as long as 4 hours.

45
Only intravenous formulations are available, because oral preparations of GP IIb/IIIa blockers are too toxic. Eptifibatide and its metabolites are excreted by the kidney. Tirofiban is excreted unchanged by the kidney. The major adverse effect of both drugs is bleeding.

48
Dipyridamole Dipyridamole [dye-peer-ID-a-mole], a coronary vasodilator, is employed prophylactically to treat angina pectoris. It is usually given in combination with aspirin or warfarin it is ineffective when used alone. Dipyridamole increases intracellular levels of cAMP by inhibiting cyclic nucleotide phosphodiesterase.
![Dipyridamole Dipyridamole [dye-peer-ID-a-mole], a coronary vasodilator, is employed prophylactically to treat angina pectoris.](http://slideplayer.com/slide/4494387/14/images/48/Dipyridamole+Dipyridamole+%5Bdye-peer-ID-a-mole%5D%2C+a+coronary+vasodilator%2C+is+employed+prophylactically+to+treat+angina+pectoris..jpg "It is usually given in combination with aspirin or warfarin. it is ineffective when used alone. Dipyridamole increases intracellular levels of cAMP by inhibiting cyclic nucleotide phosphodiesterase.")
49
Sites of Action of Antiplatelet Therapy on Mechanisms of Platelet Activation and Aggregation
Schulman, S. P. JAMA 2004;292: Copyright restrictions may apply.

50
Thrombosis - Pathogenesis
3 primary influences predispose to thrombus formation Virchow’s Triad (1856): Endothelial Injury Stasis Hypercoagulability
: Endothelial Injury. Stasis. Hypercoagulability.")
51
Venous Thrombi Most occur in the superficial or deep veins of the leg (DVT) Superficial thrombi Swelling and pain Rarely embolize DVT Pain, redness and swelling Asymptomatic in 50% Risk of emboli

52
Pathophysiology of Pulmonary Embolism
Figure 1. Pathophysiology of Pulmonary Embolism. Pulmonary embolism usually originates from the deep veins of the legs, most commonly the calf veins. These venous thrombi originate predominantly in venous valve pockets and at other sites of presumed venous stasis (inset, bottom). If a clot propagates to the knee vein or above, or if it originates above the knee, the risk of embolism increases. Thromboemboli travel through the right side of the heart to reach the lungs. LA denotes left atrium, LV left ventricle, RA right atrium, and RV right ventricle. Tapson V. N Engl J Med 2008;358:
. If a clot propagates to the knee vein or above, or if it originates above the knee, the risk of embolism increases. Thromboemboli travel through the right side of the heart to reach the lungs. LA denotes left atrium, LV left ventricle, RA right atrium, and RV right ventricle. Tapson V. N Engl J Med 2008;358:")
54
anticoagulants

55
Coagulation Phase Two major pathways Both converge at a common point
Intrinsic pathway Extrinsic pathway Both converge at a common point Biosynthesis of these factors are dependent on Vitamin K1 and K2 Normally inactive and sequentially activated

56
Intrinsic Pathway All clotting factors are within the blood vessels Clotting slower Activated partial thromboplastin test (aPTT) Extrinsic Pathway Initiating factor is outside the blood vessels - tissue factor Clotting - faster - in Seconds Prothrombin test (PT)
")
57
Intrinsic Pathway Extrinsic Pathway Blood Vessel Injury Tissue Injury
Tissue Factor XII XIIa Thromboplastin XI XIa IX IXa VIIa VII X Xa X Prothrombin Thrombin Factors affected By Heparin Fibrinogen Fribrin monomer Fibrin polymer Vit. K dependent Factors Affected by Oral Anticoagulants

58
The Coagulation and Fibrinolytic Pathways
Figure 1. The Coagulation and Fibrinolytic Pathways. The main coagulation reactions are divided into the intrinsic and extrinsic systems. Activation of factor XII on contact with a negatively charged surface initiates the intrinsic coagulation system. (The activated form of the factor is indicated by "a.") The extrinsic coagulation system induces the formation of a complex composed of factor VII and tissue factor, which is released after tissue injury. Some of these reactions depend on calcium ions. Thrombin is formed by an enzyme complex called prothrombinase, composed of factor X, factor V, negatively charged phospholipids, and calcium ions. Intrinsic and extrinsic activation of the coagulation cascade leads to the generation of thrombin, the activation of fibrinogen, the release of fibrinopeptides, the formation of soluble fibrin, and finally, the formation of insoluble fibrin. The main fibrinolytic reactions involve the inhibition of fibrinolysis by plasminogen-activator inhibitor type 1 (PAI-1) and {alpha}2-antiplasmin. Fibrinolysis is initiated by tissue plasminogen activator (t-PA), urinary-type plasminogen activator (u-PA), and plasmin. Plasmin bound to the surface of fibrin initiates the lysis of insoluble, cross-linked fibrin, with the subsequent generation of fibrin-degradation products. Plasmin bound to the surface of fibrin is better protected from inhibition by {alpha}2-antiplasmin than is plasmin generated in the fluid phase.
 The extrinsic coagulation system induces the formation of a complex composed of factor VII and tissue factor, which is released after tissue injury. Some of these reactions depend on calcium ions. Thrombin is formed by an enzyme complex called prothrombinase, composed of factor X, factor V, negatively charged phospholipids, and calcium ions. Intrinsic and extrinsic activation of the coagulation cascade leads to the generation of thrombin, the activation of fibrinogen, the release of fibrinopeptides, the formation of soluble fibrin, and finally, the formation of insoluble fibrin. The main fibrinolytic reactions involve the inhibition of fibrinolysis by plasminogen-activator inhibitor type 1 (PAI-1) and {alpha}2-antiplasmin. Fibrinolysis is initiated by tissue plasminogen activator (t-PA), urinary-type plasminogen activator (u-PA), and plasmin. Plasmin bound to the surface of fibrin initiates the lysis of insoluble, cross-linked fibrin, with the subsequent generation of fibrin-degradation products. Plasmin bound to the surface of fibrin is better protected from inhibition by {alpha}2-antiplasmin than is plasmin generated in the fluid phase.")
59
Coagulation cascade Prothrombin thrombin
Present on platelets’ surfaces. Act by accelerating thrombus formation. Coagulation cascade Activated factor X (FXa) + FVa + Ca++ + phospholipids Prothrombin thrombin Fibrinogen fibrin blood clot
 + FVa + Ca++ + phospholipids. Prothrombin thrombin. Fibrinogen fibrin blood clot.")
60
Platelets aggregation ↔ coagulation
This complex and Ca2+ comprise the prothrombinase complex Thrombin stimulates platelet aggregation Coagulation ↑ platelet aggregation Click to add note.. Phospholipids on platelets stimulate clot formation Platelets aggregation ↔ coagulation

61
The coagulation process that generates thrombin consists of two interrelated pathways the extrinsic and the intrinsic systems. The extrinsic system, which is probably the more important system in vivo, is initiated by the activation of clotting Factor VII by tissue factor, or thromboplastin. Tissue factor is a lipoprotein that is expressed by cells at the site of vascular injury.

62
The intrinsic system is triggered by the activation of clotting Factors its contact in vitro with glass or highly charged surfaces. Intrinsic and extrinsic activation of the coagulation cascade leads to the generation of thrombin, the activation of fibrinogen, the release of fibrinopeptides, the formation of soluble fibrin, and finally, the formation of cross-linked, insoluble fibrin. (The activated form of the factor is indicated by "a.")
")
63
It is important that coagulation is restricted to the local site of vascular injury.
Endogenously, there are several inhibitors of coagulation factors, including protein C, protein S, antithrombin III, and tissue factor pathway inhibitor. The mechanism of action of several anticoagulant agents, including heparin and heparin-related products, involves activation of these endogenous inhibitors (primarily antithrombin III).
.")
64
anticoagulants The anticoagulant drugs either:
inhibit the action of the coagulation factors (the thrombin inhibitors, such as heparin and heparin-related agents) interfere with the synthesis of the coagulation factors (the vitamin K antagonists, such as warfarin).
 interfere with the synthesis of the coagulation factors (the vitamin K antagonists, such as warfarin).")
66
Hemostasis requires a fine balance between
procoagulant and regulatory factors Coagulation Proteins/ Platelets/ Vessel wall PC PS ATIII… Thrombosis Deficiency Deficiency/ Abnormality Bleeding

67
Anticoagulants FACTORY

68
Scheme of anticoagulant drugs
oral warfarin injected Direct acting: Lepirudin Antithrombin III dependent: heparin Low molecular weight heparin

69
Injected; THROMBIN INHIBITORS
Thrombin inhibitors can either inactivate thrombin directly or block thrombin formation Thrombin can be inhibited irreversibly by glycosaminoglycans like heparin through an antithrombin III-dependent mechanism The enzyme can be inhibited reversibly by hirudin and hirudin derivatives in an antithrombin III-independent manner (direct acting) In addition to inhibiting thrombin, glycosaminoglycans also block thrombin generation
 In addition to inhibiting thrombin, glycosaminoglycans also block thrombin generation.")
70
Antithrombin-III Dependent Thrombin Inhibitors
Standard Unfractionated Heparin (UFH) Heparin is a mixture of glycosaminoglycan molecules, which are heterogenous in molecular size The mean molecular weight of heparin is 15,000 D Antithrombin III (ATIII) binding is necessary for its anticoagulant activity Antithrombin III (ATIII) is a slow endogenous progressive inhibitor of thrombin and other clotting enzymes (Xa)
 Heparin is a mixture of glycosaminoglycan molecules, which are heterogenous in molecular size. The mean molecular weight of heparin is 15,000 D. Antithrombin III (ATIII) binding is necessary for its anticoagulant activity. Antithrombin III (ATIII) is a slow endogenous progressive inhibitor of thrombin and other clotting enzymes (Xa)")
71
Mode of Action of Heparin
It has 2 mechanisms It binds to ATIII through a unique pentasaccharide conformational change in ATIII ↑ activity of ATIII Heparin acts as a template to create (thrombin-ATIII complex) N.B. only 1 ATIII bind to 1 thrombin (1:1) N.B. ATIII alone can inhibit thrombin but in a very slow reaction Then heparin dissociates and is reused again
 N.B. only 1 ATIII bind to 1 thrombin (1:1) N.B. ATIII alone can inhibit thrombin but in a very slow reaction. Then heparin dissociates and is reused again.")
72
Heparin inactivates thrombin by binding both ATIII and thrombin
To inactivate thrombin Heparin binds to ATIII by the unique penta-saccharide Also binds to thrombin through the heparin-binding domain heparn also inactivate factor Xa, but in such case it binds only with ATIII through its pentasaccharide sequence Every heparin molecule contains : Pentasaccharide + heparin binding domain Anti-IIa activity = Anti-Xa activity

75
Low Molecular Weight Heparins (LMWHs)
Low molecular weight heparin have a mean molecular weight of 5000 D. Prepared by controlled chemical or enzymatic depolymerization of standard unfractioned heparin are about ⅓ the size of starting material Enoxaparin is the most used LMWH

76
Mechanism of Action of Low Molecular Weight Heparin (LMWH)
They contain pentasaccharide inactivation of Factor Xa In contrast, only 25% to 50% of LMWH molecules that have the pentasaccharide sequence are long enough to interact with both ATIII & thrombin fix 25% of LMWH can interact with both ATIII and thrombin The rest only inactivate factor X Anti-IIa < Anti-Xa activity

77
Pharmacokinetic Profile of LMWH
Bioavilability Bbioavailability (90% vs. 20% of heparin) LMWH exhibit less binding to plasma proteins & cell surfaces (better than heparin) more predictable anticoagulant response Laboratory monitoring of LMWH activity is not required LMWH has low resistance in comparison to heparin T1/2 = 4 hours (more than heparin) Given at fixed doses once to twice daily by S.C. route, and is given for both inpatients as well as for outpatients.
 LMWH exhibit less binding to plasma proteins & cell surfaces (better than heparin) more predictable anticoagulant response. Laboratory monitoring of LMWH activity is not required. LMWH has low resistance in comparison to heparin. T1/2 = 4 hours (more than heparin) Given at fixed doses once to twice daily by S.C. route, and is given for both inpatients as well as for outpatients.")
78
Comparison of UFH & LMWH
Character UFH LMWH Average Mol wt 15,000 5,000 Anti-Xa/anti-IIa activity 1/1 2-4/1 aPTT monitoring required Yes No Inactivation of platelet-bound Xa Protein binding Powerful) 4+) Weak (+) Endothelial cell binding Dose-dependent clearance Elimination half-life min 2-5 times longer
 4+) Weak (+) Endothelial cell binding. Dose-dependent clearance. Elimination half-life min. 2-5 times longer.")
79
Biophysical Limitations of Heparin and LMWH
Both heparin and LMWH can’t degrade fibrin-bound thrombin (only free thrombin is degraded) nor Factor Xa within the prothrombinase complex.
 nor Factor Xa within the prothrombinase complex.")
80
Therapeutic Uses Heparin should be given either IV or S.C. injection.
onset of action: few minutes (IV) 1-2 hours (S.C.) LMWHs are given by S.C. route I.M. injection hematoma formation (thus is avoided) Treatment of deep vein thrombosis Treatment of pulmonary embolism Prevention of postoperative venous thrombosis in patients with acute MI phase or one undergoing elective surgery (not emergency surgery) Reduction of coronary artery thrombosis after thrombolytic treatment Heparin is the anticoagulant of choice in pregnant women Elective surgery? Ya’ny not an emergency operation.
 1-2 hours (S.C.) LMWHs are given by S.C. route. I.M. injection hematoma formation (thus is avoided) Treatment of deep vein thrombosis. Treatment of pulmonary embolism. Prevention of postoperative venous thrombosis in patients with acute MI phase or one undergoing elective surgery (not emergency surgery) Reduction of coronary artery thrombosis after thrombolytic treatment. Heparin is the anticoagulant of choice in pregnant women. Elective surgery Ya’ny not an emergency operation.")
81
Heparin and LMWHs are the anticoagulants of choice for treating pregnant women with prosthetic heart valves or venous thromboembolism, because these agents do not cross the placenta (due to their large size and negative charge). Heparin has the advantage of speedy onset of action, which is rapidly terminated on suspension of therapy. However, it is being supplanted by the LMWHs, such as enoxaparin and dalteparin, because they can be conveniently injected subcutaneously on a patient weight basis, have predictable therapeutic effects, and have a more predictable pharmacokinetic profile

82
Adverse Effects Bleeding: they both lead to bleeding but the bleeding is less in LMWH To treat bleeding: inject antidote protamine sulphate (1mg IV for each 100 units of UFH) (reversal effect) Thrombosis and heparin induced Thrombocytopenia (HI)T: HIT is caused by the formation of abnormal antibodies that activate platelets. so someone receiving heparin develops new or worsening thrombosis, or if the platelet count falls.
 (reversal effect) Thrombosis and heparin induced Thrombocytopenia (HI)T: HIT is caused by the formation of abnormal antibodies that activate platelets. so someone receiving heparin develops new or worsening thrombosis, or if the platelet count falls.")
83
For patients with HIT we use lepirudin
HIT is a life threatening immune reaction Occurs in 3% of patients Usually occurs a week from starting heparin therapy LMWHs, though of lower risk, are contraindicated with HIT. For patients with HIT we use lepirudin

84
How does HIT occur? Heparin injection immune reaction with body produce antibody against heparin & also bind to platelet receptor activation of platelet thrombosis

85
Osteoporosis occurs with large doses of UFH >20,000 U/day for 6 months or longer (chronic use)
Hyperkalemia rarely occurs with UFH It is attributed to inhibition of aldosterone secretion It is reversible by therapy discontinuation Diabetic & renal failure patients are at higher risk Hypersensitivity: (Antigenicity due to animal source) rarely occurring reactions include urticaria, rash, rhinitis, angioedema & reversible alopecia(hair loss)
 rarely occurring reactions include urticaria, rash, rhinitis, angioedema & reversible alopecia(hair loss)")
86
Thrombocytopenia or purpura Hypersensitivity to heparin
contraindications Bleeding or hemophilia hypertension Thrombocytopenia or purpura Hypersensitivity to heparin Intracranial hemorrhage Recent surgery TB GIT ulcer Hepatic or renal disease Use of digoxin fix

87
HEPARIN TEST of CONTROL
ACTIVATED PARTIALTHROMBOPLASTIN TIME OR aPTT or PTT A THERAPEUTIC VALUE, ~ 0.3 u/ml REVERSAL UFH – Protamine LMWH – Protamine not fully effective

89
An 83-year-old woman with a history of hypertension, glaucoma, and atrial fibrillation, who had a pacemaker and was receiving warfarin, woke up with a sensation of pain in the right eye and noticed what she thought was a big bruise. She had no headache, trauma, nuchal rigidity, nausea, or photophobia. Physical examination showed a subconjunctival hemorrhage in the right eye, as well as a defect in the nasal and superior temporal visual field, periorbital ecchymosis, and edema. The patient's visual acuity was unchanged from baseline. The blood pressure was 192/90 mm Hg, the intraocular pressure was 20 mm Hg, the hemoglobin level was 14.6 g per deciliter, and the international normalized ratio (INR) was 2.2. Computed tomography showed modest hemorrhage in the preseptal tissues overlying the globe and complete opacification of the right maxillary sinus with blood but no intracerebral hemorrhage or fracture. The bleeding slowly resolved over a period of six weeks. Because the INR was in the range desired, the dose of warfarin was not changed. There were no subsequent visual-field defects. The patient was not referred for further treatment and had no subsequent episodes. Stead L and Judson K. N Engl J Med 2006;355:e7

90
Other Injectable Antithrombotic Agents
Fondaparinux, a pentasaccharide, is an AT-III-dependent selective for factor Xa Prevents venous thrombosis associated with orthopedic surgery Administered > 6 hours postoperatively and the dose is adjusted for patients with renal impairment.

91
FONDAPARINUX

92
It is well absorbed from the subcutaneous route with a predictable pharmacokinetic profile.
Fondaparinux requires less monitoring than heparin. Fondaparinux is eliminated in urine mainly as unchanged drug with an elimination half-life of 17 to 21 hours. It is contraindicated in patients with severe renal impairment (<30 mL/min). Bleeding episodes are the major side effect of fondaparinux therapy. Thrombocytopenia, is not a problem, and this agent may be used in patients with HIT.
. Bleeding episodes are the major side effect of fondaparinux therapy. Thrombocytopenia, is not a problem, and this agent may be used in patients with HIT.")
93
Clinically Approved Direct Thrombin Inhibitors
Lepirudin, recombinant hirudin*-like peptide. Direct acting thrombin inhibitor Used in HIT patients (IV injection) Has renal clearance It acts on free thrombin and thrombin bound to fibrin It has potential use in unstable angina patients and after thrombolysis Mechanism of action * hirudin: is a leech derived anticoagulant It binds to active site and substrate site of thrombin
 Has renal clearance. It acts on free thrombin and thrombin bound to fibrin. It has potential use in unstable angina patients and after thrombolysis. Mechanism of action. * hirudin: is a leech derived anticoagulant. It binds to active site and substrate site of thrombin.")
94
Bleeding is the major adverse effect of treatment with lepirudin, and it can be exacerbated by concomitant thrombolytic therapy, such as treatment with streptokinase or alteplase. About half the patients receiving lepirudin develop antibodies. However, the drug-antibody complex retains anticoagulant activity. Because renal elimination of the complex is slower than that of the free drug, the anticoagulant effect may be increased. It is important to monitor the aPTT and renal function when a patient is receiving lepirudin.

95
Argatroban is metabolized in the liver and has a half life of about 50 minutes. It is monitored by aPTT. The patient's hemoglobin and hematocrit must also be monitored. Because argatroban is metabolized in the liver, it may be used in patients with renal dysfunction but it should be used cautiously in patients with hepatic impairment. As with other agents in this class, the major side effect is bleeding.

96
Mechanism of Action of Direct Thrombin Inhibitors as Compared with Heparin
Figure 2. Mechanism of Action of Direct Thrombin Inhibitors as Compared with Heparin. In the absence of heparin, the rate of thrombin inactivation by antithrombin is relatively low, but after conformational change induced by heparin, antithrombin irreversibly binds to and inhibits the active site of thrombin. Thus, the anticoagulant activity of heparin originates from its ability to generate a ternary heparin-thrombin-antithrombin complex. The activity of DTIs is independent of the presence of antithrombin and is related to the direct interaction of these drugs with the thrombin molecule. Although bivalent DTIs simultaneously bind the exosite 1 and the active site, the univalent drugs in this class interact only with an active site of the enzyme. In the lower panel, the heparin-antithrombin complex cannot bind fibrin-bound thrombin, whereas given their mechanism of action, DTIs can bind to and inhibit the activity of not only soluble thrombin but also thrombin bound to fibrin, as is the case in a blood clot. An animated version of this figure is available with the full text of the article at Di Nisio, M. et al. N Engl J Med 2005;353:

97
Synthesis of clotting factors

98
Oral Anticoagulants Vitamin K Antagonists (The Coumarins)
Vitamin K is co-factor for the hepatic synthesis of clotting factors II, VII, IX & X warfarin By Vit.k reductase Vit. K Vit. K epoxide (active form) Warfarin inhibits Vit. K reductase no active form of Vit. K no synthesis of clotting factors
 Warfarin inhibits Vit. K reductase no active form of Vit. K no synthesis of clotting factors.")
100
ROLE of VITAMIN K

102
Vitamin K Antagonists (Warfarin)
Onset: Clinical anticoagulant activity needs several days to develop (due to the already circulating clotting factors) So the action of warfarin will appear after the elimination of prior clotting factors. Elimination time (factor II needs: 60 hours, factor X: 40 hours) lasts for 4-5 days Overlap heparin & warfarin therapy taken together until the effect of warfarin appears (after 5 days) then stop taking heparin.
 So the action of warfarin will appear after the elimination of prior clotting factors. Elimination time (factor II needs: 60 hours, factor X: 40 hours) lasts for 4-5 days. Overlap heparin & warfarin therapy taken together until the effect of warfarin appears (after 5 days) then stop taking heparin.")
103
Vitamin K Antagonists Warfarin
Warfarin has 100% oral bioavailability high plasma protein binding Warfarin is metabolized by hepatic Cytochrome P450 enzymes long plasma t1/2 of 36 hours A high loading dose followed by an adjusted maintenance dose Warfarin is contraindicated with pregnancy as it crosses the placental barrier and is teratogenic in the first trimester & and induce intracranial hemorrhage in the baby during delivery

104
Prothrombin time, a measure of the extrinsic pathway, may be used to monitor warfarin therapy.
In the 1990s, the international normalized ratio (INR) was adopted to monitor warfarin concentration. The INR corrects for variations that would occur with different thromboplastin reagents, between different hospitals, or when a single hospital gets a new lot of reagent. The goal of warfarin therapy is an INR of 2 to 3 for most indications and 2.5 to 3.5 in patients with mechanical heart valves.
 was adopted to monitor warfarin concentration. The INR corrects for variations that would occur with different thromboplastin reagents, between different hospitals, or when a single hospital gets a new lot of reagent. The goal of warfarin therapy is an INR of 2 to 3 for most indications and 2.5 to 3.5 in patients with mechanical heart valves.")
105
Warfarin is used to prevent the progression or recurrence of acute deep-vein thrombosis or pulmonary embolism after initial heparin treatment. It is also used for the prevention of venous thromboembolism during orthopaedic or gynecologic surgery. Prophylactically, it is used in patients with acute myocardial infarction, prosthetic heart valves, or chronic atrial fibrillation.

106
Warfarin Drug Pharmacokinetic & Pharmacodynamic Interactions
Potentiating warfarin Inhibitors of hepatic P450 enzymes (cimetidine, cotrimoxazole, imipramine, amiodarone) Platelet aggregation inhibitors (NSAIDs e.g. aspirin) 3rd generation cephalosporins* Drugs displacing warfarin from binding sites (NSAIDs) Drugs reducing the availability of vitamin K Hepatic disease(↓ clotting factors) & hyperthyroidism Inhibiting Warfarin Vitamin K Inducers of hepatic P450 enzymes (rifampicin, barbiturates, … etc) Reduction of GIT absorption (cholestyramine) Diuretics Hypothyroidism *Cephalosporins potentiate warfarin’s effect by killing vit.k producing normal flora
 Platelet aggregation inhibitors (NSAIDs e.g. aspirin) 3rd generation cephalosporins* Drugs displacing warfarin from binding sites (NSAIDs) Drugs reducing the availability of vitamin K. Hepatic disease(↓ clotting factors) & hyperthyroidism. Inhibiting Warfarin. Vitamin K. Inducers of hepatic P450 enzymes (rifampicin, barbiturates, … etc) Reduction of GIT absorption (cholestyramine) Diuretics. Hypothyroidism. *Cephalosporins potentiate warfarin’s effect by killing vit.k producing normal flora.")
107
Warfarin Side-Effects
Drug-drug interactions Bleeding disorder (thus should be monitored) Treatment for bleeding Minor bleeding: stop therapy + oral Vitamin K Severe Bleeding: stop therapy + I.V. Vitamin K fresh frozen plasma, recombinant factor VIIa or prothrombin complex may be used
 Treatment for bleeding. Minor bleeding: stop therapy + oral Vitamin K. Severe Bleeding: stop therapy + I.V. Vitamin K fresh frozen plasma, recombinant factor VIIa or prothrombin complex may be used.")
108
WARFARIN – DRUG INTERACTIONS
Diminished warfarin actions Ethanol Enhanced warfarin actions Ethanol Aspirin Cimetidine

109
Thrombolytics

112
Fibrinolysis is initiated by tissue plasminogen activator (t-PA), urinary-type plasminogen activator (u-PA), and plasmin. Plasmin bound to the surface of fibrin initiates the lysis of insoluble, cross-linked fibrin, with the subsequent generation of fibrin-degradation products. The main fibrinolytic reactions involve the inhibition of fibrinolysis by plasminogen-activator inhibitor type 1 (PAI-1) and {α}2-antiplasmin.
 and {α}2-antiplasmin.")
113
The balance between the formation and degradation of FN
Nesheim, M. Chest 2003;124:33S-39S

115
Thrmobolytics Acute thromboembolic disease in selected patients may be treated by the administration of agents that activate the conversion of plasminogen to plasmin that hydrolyzes fibrin and, thus, dissolves clots.

116
Streptokinase, one of the first such agents to be approved, causes a systemic fibrinolytic state that can lead to bleeding problems. Alteplase acts more locally on the thrombotic fibrin to produce fibrinolysis.. Clinical experience has shown nearly equal efficacy betweenn streptokinase and alteplase..

117
Plasmin bound to the surface of fibrin is better protected from inhibition by {α}2-antiplasmin than is plasmin generated in the fluid phase.

118
thrombolytic therapy is unsuccessful in about 20 % of infarcted arteries, and about 15 % of the arteries that are opened will later close again. In the case of acute myocardial infarction, the thrombolytic drugs are reserved for those instances when angioplasty is not an option or until the patient can be taken to a facility that performs percutaneous coronary interventions.

119
Clot dissolution and reperfusion occur with a higher frequency when therapy is initiated early after clot formation, because clots become more resistant to lysis as they age. Fibrinolytic drugs may lyse both normal and pathologic thrombi.

122
thrombolytic agents are helpful in restoring catheter and shunt function, by lysing clots causing occlusions. Thrombolytic agents are also used to dissolve clots that result in strokes. thrombolytic agents are usually administered intravenously. These drugs are contraindicated in patients with healing wounds, pregnancy, history of cerebrovascular accident, or metastatic cancer.

123
streptokinase Mechanism of action:
Streptokinase has no enzymic activity. Instead, it forms an active one-to-one complex with plasminogen. This enzymatically active complex converts uncomplexed plasminogen to the active enzyme plasmin. In addition to the hydrolysis of fibrin plugs, the complex also catalyzes the degradation of fibrinogen as well as clotting Factors V and VII.

126
Therapeutic uses: Streptokinase is approved for use in acute pulmonary embolism, deep-vein thrombosis, acute myocardial infarction, arterial thrombosis, and occluded access shunts. Pharmacokinetics: Streptokinase therapy is instituted within 4 hours of a myocardial infarction and is infused for 1 hour. Its half-life is less than half an hour.

127
Thromboplastin time is monitored and maintained at two- to five-fold the control value.
On discontinuation of treatment, either heparin or oral anticoagulants may be administered.

129
Hypersensitivity: Streptokinase is a foreign protein and is antigenic. Rashes, fever, and rarely, anaphylaxis occur. Because most individuals have had a streptococcal infection sometime in their lives, circulating antibodies against streptokinase are likely to be present in most patients. These antibodies can combine with streptokinase and neutralize its fibrinolytic properties. Therefore, sufficient quantities of streptokinase must be administered to overwhelm the antibodies and provide a therapeutic concentration of plasmin. The incidence of allergic reactions is approximately 3 percent.

131
alteplase Alteplase [AL-te-place] (formerly known as tissue plasminogen activator, or tPA) is a serine protease originally derived from cultured human melanoma cells.
 is a serine protease originally derived from cultured human melanoma cells.")
132
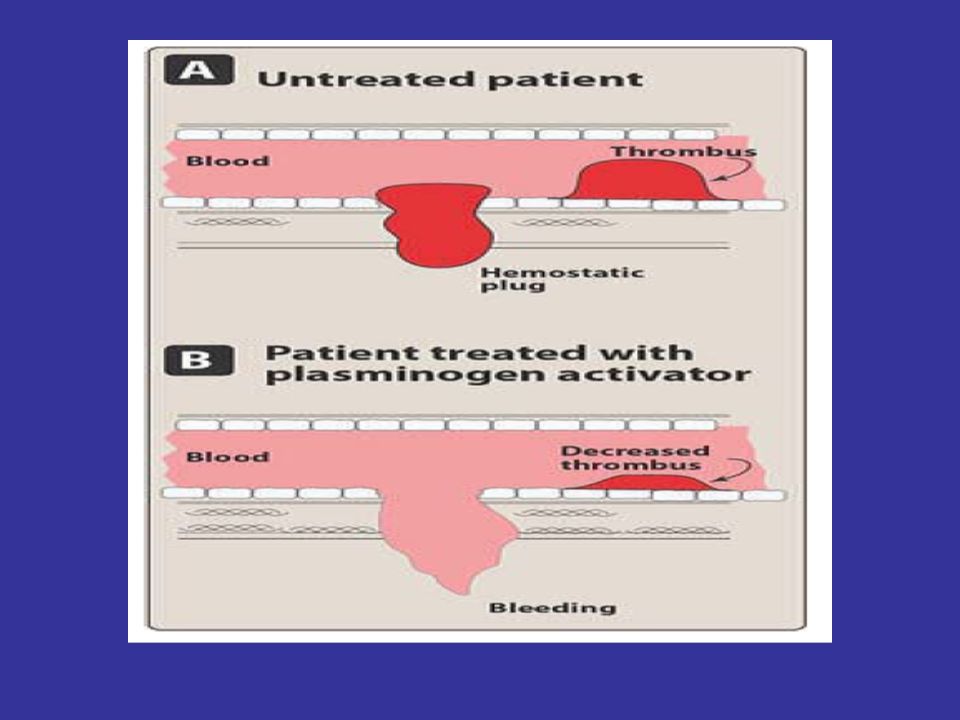
Mechanism of action: Alteplase has a low affinity for free plasminogen in the plasma, but it rapidly activates plasminogen that is bound to fibrin in a thrombus or a hemostatic plug. Thus, alteplase is said to be fibrin selective, and at low doses, it has the advantage of lysing only fibrin, without unwanted degradation of other proteins notably fibrinogen. This contrasts with streptokinase, which acts on free plasminogen and induces a general fibrinolytic state.

134
Alteplase seems to be superior to streptokinase in dissolving older clots.
Alteplase, administered within 3 hours of the onset of ischemic stroke, significantly improves clinical outcome that is, the patient's ability to perform activities of daily living. Reteplase (Retavase) is similar to alteplase and can be used as an alternative.
 is similar to alteplase and can be used as an alternative.")
136
Anistreplase (plasminogen streptokinase activator complex)
Anistreplase is a preformed complex of streptokinase and plasminogen and it is considered to be a prodrug.

137
Agents that control bleeding

138
Drugs Used to Treat Bleeding
A. Aminocaproic acid and tranexamic acid Both agents are synthetic, inhibit plasminogen activation, are orally active, and are excreted in the urine. A potential side effect of treatment is intravascular thrombosis.

139
Protamine sulfate antagonizes the anticoagulant effects of heparin. Adverse effects of drug administration include hypersensitivity as well as dyspnea, flushing, bradycardia, and hypotension when rapidly injected.

140
Vitamin K That vitamin K1 (phytonadione) administration can stop bleeding problems due to the oral anticoagulants is not surprising, because those substances act by interfering with the action of the vitamin. The response to vitamin K is slow, requiring about 24 hours (time to synthesize new coagulation factors). Thus, if immediate hemostasis is required, fresh-frozen plasma should be infused.
 administration can stop bleeding problems due to the oral anticoagulants is not surprising, because those substances act by interfering with the action of the vitamin. The response to vitamin K is slow, requiring about 24 hours (time to synthesize new coagulation factors). Thus, if immediate hemostasis is required, fresh-frozen plasma should be infused.")
141
Aprotinin stops bleeding by blocking plasmin.
It can inhibit streptokinase. It is approved for prophylactic use to reduce perioperative blood loss and the need for blood transfusion in patients undergoing cardiopulmonary bypass surgery. Aprotinin may cause renal dysfunction and hypersensitivity (anaphylactic) reactions. In addition, aprotinin should not be administered to patients who have already been exposed to the drug within the previous 1 year due to the possibility of anaphylactic reactions.
 reactions. In addition, aprotinin should not be administered to patients who have already been exposed to the drug within the previous 1 year due to the possibility of anaphylactic reactions.")
142
What is the next topic?

143
Anti Anemic Drugs Anti anaemic Drugs (1).ppt
.ppt")
Similar presentations





























>")


 and prostacyclinre (PGI2)leased.>")






 By Prof. Hanan Hagar Dr.Abdul latif Mahesar 1.>")

 and Clopidogrel (Plavix™) Benedict R. Lucchesi, M.D., Ph.D. Department of Pharmacology University of Michigan Medical School.>")

