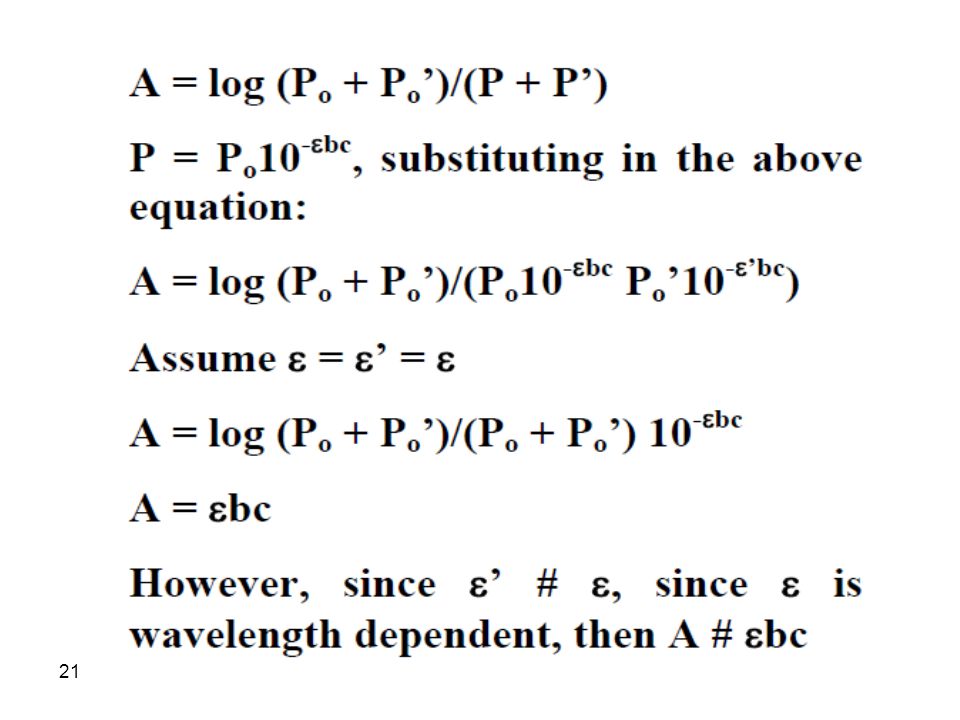Download presentation
Presentation is loading. Please wait.

1
An introduction to Ultraviolet/Visible Absorption Spectroscopy
Chapter 13

2
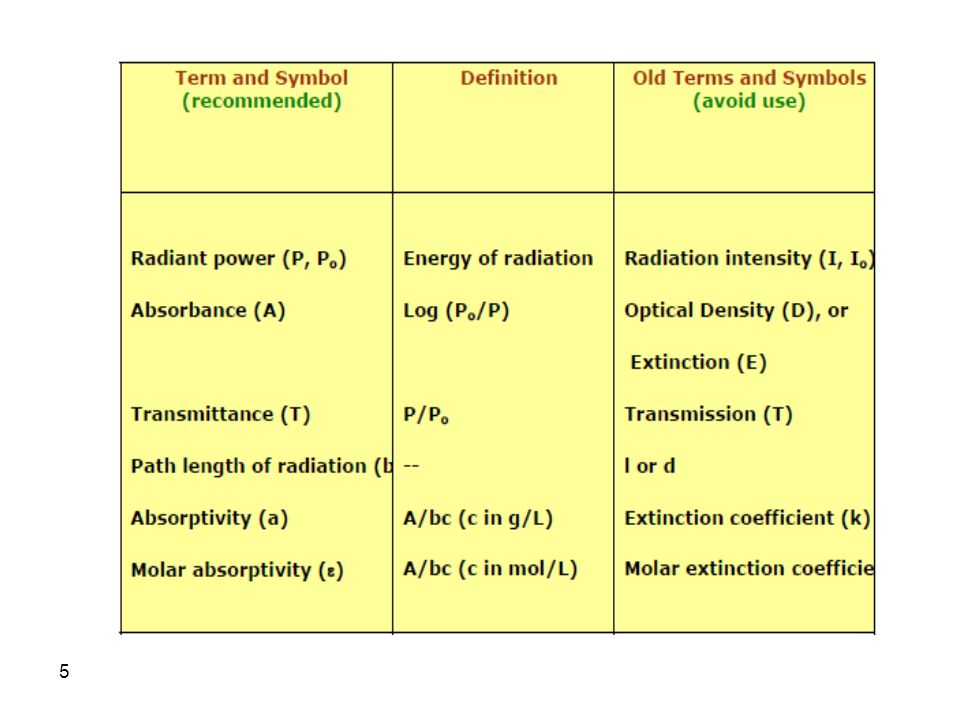
In this chapter, absorption by molecules, rather than atoms, is considered. Absorption in the ultraviolet and visible regions occurs due to electronic transitions from the ground state to excited state. Broad band spectra are obtained since molecules have vibrational and rotational energy levels associated with electronic energy levels. The signal is either absorbance or percent transmittance of the analyte solution where:

3
An Introduction to Ultraviolet/Visible Molecular Absorption Spectrometry
Absorption measurements based upon ultraviolet and visible radiation find widespread application for the quantitative determination of a large variety species. Beer’s Law: A = -logT = logP0/P = bc A = absorbance = molar absorptivity [M-1 cm-1] c = concentration [M] P0 = incident power P = transmitted power (after passing through sample)
")
7
Measurement of Transmittance and Absorbance:
The power of the beam transmitted by the analyte solution is usually compared with the power of the beam transmitted by an identical cell containing only solvent. An experimental transmittance and absorbance are then obtained with the equations. P0 and P refers to the power of radiation after it has passed through the solvent and the analyte.

8
Beer’s law and mixtures
Each analyte present in the solution absorbs light! The magnitude of the absorption depends on its e A total = A1+A2+…+An A total = e1bc1+e2bc2+…+enbcn If e1 = e2 = en then simultaneous determination is impossible Need nl’s where e’s are different to solve the mixture

9
Limitations to Beer’s Law
Real limitations Chemical deviations Instrumental deviations

10
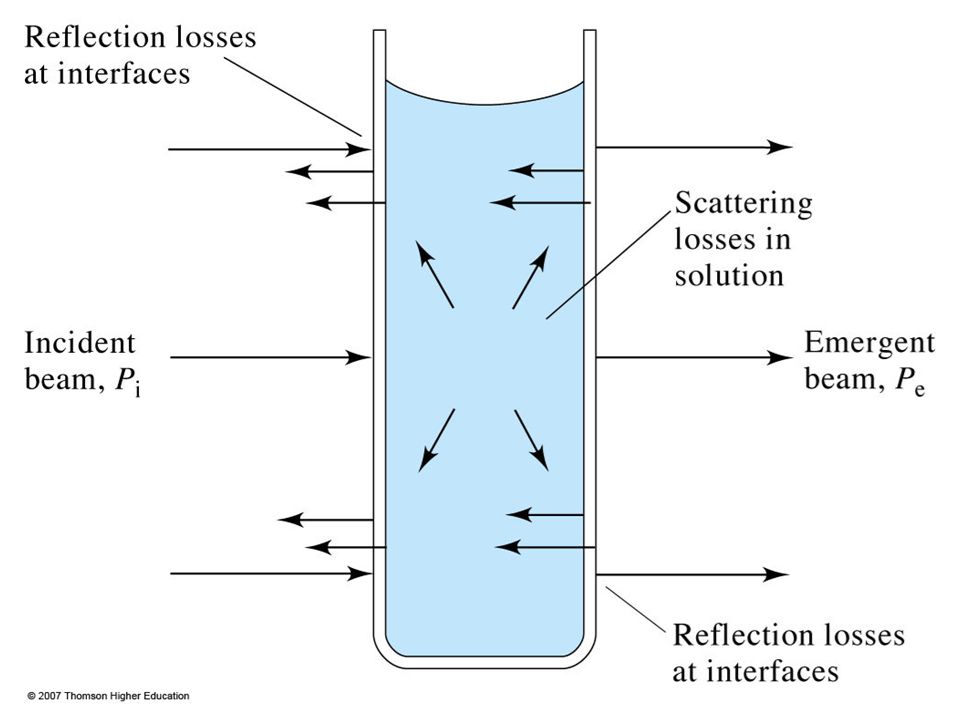
1. Real Limitations a. Beer’s law is good for dilute analyte solutions only. High concentrations (>0.01M) will cause a negative error since as the distance between molecules become smaller the charge distribution will be affected which alter the molecules ability to absorb a specific wavelength. The same phenomenon is also observed for solutions with high electrolyte concentration, even at low analyte concentration. The molar absorptivity is altered due to electrostatic interactions.
 will cause a negative error since as the distance between molecules become smaller the charge distribution will be affected which alter the molecules ability to absorb a specific wavelength. The same phenomenon is also observed for solutions with high electrolyte concentration, even at low analyte concentration. The molar absorptivity is altered due to electrostatic interactions.")
11
b. In the derivation of Beer’s law we have introduced a constant (e)
b. In the derivation of Beer’s law we have introduced a constant (e). However, e is dependent on the refractive index and the refractive index is a function of concentration. Therefore, e will be concentration dependent. However, the refractive index changes very slightly for dilute solutions and thus we can practically assume that e is constant. c. In rare cases, the molar absorptivity changes widely with concentration, even at dilute solutions. Therefore, Beer’s law is never a linear relation for such compounds, like methylene blue.
. However, e is dependent on the refractive index and the refractive index is a function of concentration. Therefore, e will be concentration dependent. However, the refractive index changes very slightly for dilute solutions and thus we can practically assume that e is constant. c. In rare cases, the molar absorptivity changes widely with concentration, even at dilute solutions. Therefore, Beer’s law is never a linear relation for such compounds, like methylene blue.")
12
2. Chemical Deviations This factor is an important one which largely affects linearity in Beer’s law. It originates when an analyte dissociates, associates, or reacts in the solvent. For example, an acid base indicator when dissolved in water will partially dissociate according to its acid dissociation constant:

17
Chemical deviations from Beer’s law for unbuffered solutions of the indicator Hln. Note that there are positive deviations at 430 nm and negative deviations at 570 nm. At 430 nm, the absorbance is primarily due to the ionized In- form of the indicator and is proportional to the fraction ionized, which varies nonlinearly with the total indicator concentration. At 570 nm, the absorbance is due principally to the undissociated acid Hln, which increases nonlinearly with the total concentration.

18
Calculated Absorbance Data for Various Indicator Concentrations

20
3. Instrumental Deviations
a. Beer’s law is good for monochromatic light only since e is wavelength dependent. It is enough to assume a dichromatic beam passing through a sample to appreciate the need for a monochromatic light. Assume that the radiant power of incident radiation is Po and Po’ while transmitted power is P and P’. The absorbance of solution can be written as:

23
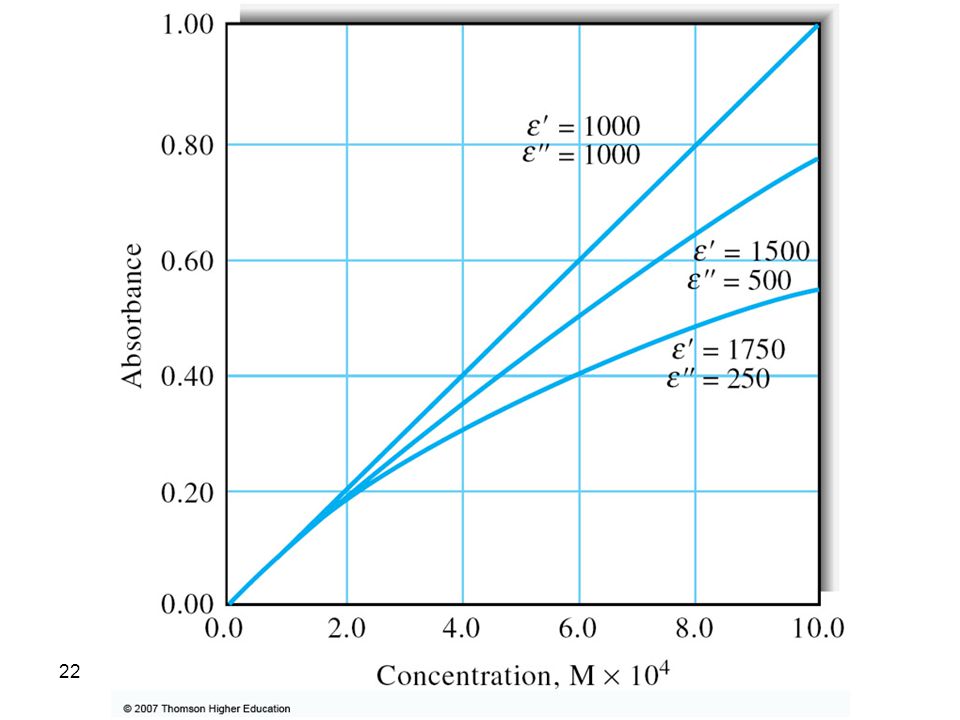
The effect of polychromatic radiation on Beer’s law
The effect of polychromatic radiation on Beer’s law. In the spectrum at the top, the absorptivity of the analyte is nearly constant over Band A from the source. Note in the Beer’s law plot at the bottom that using Band A gives a linear relationship. In the spectrum, Band B corresponds to a region where the absorptivity shows substantial changes. In the lower plot, note the dramatic deviation from Beer’s law that results.

25
Therefore, the linearity between absorbance and concentration breaks down if incident radiation was polychromatic. In most cases with UV-Vis spectroscopy, the effect small changes in wavelengths is insignificant since e differs only slightly; especially at the wavelength maximum.

26
b. Stray Radiation Stray radiation resulting from scattering or various reflections in the instrument will reach the detector without passing through the sample. The problem can be severe in cases of high absorbance or when the wavelengths of stray radiation is in such a range where the detector is highly sensitive as well as at wavelengths extremes of an instrument. The absorbance recorded can be represented by the relation: A = log (Po + Ps)/(P + Ps) Where; Ps is the radiant power of stray radiation.
/(P + Ps) Where; Ps is the radiant power of stray radiation.")
28
Instrumental Noise as a Function in Transmittance

32
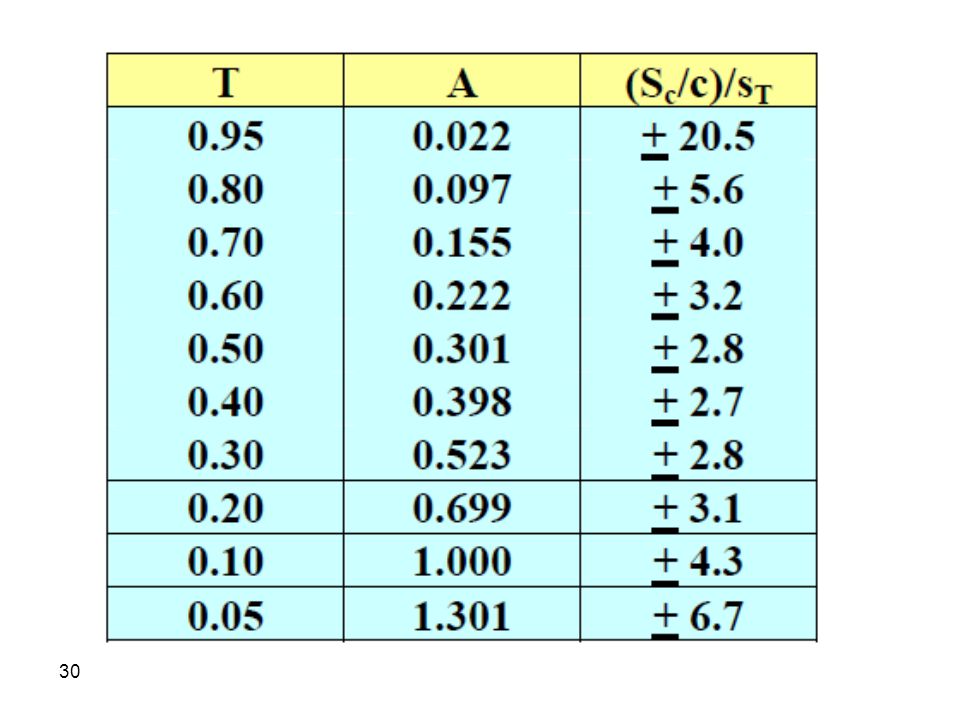
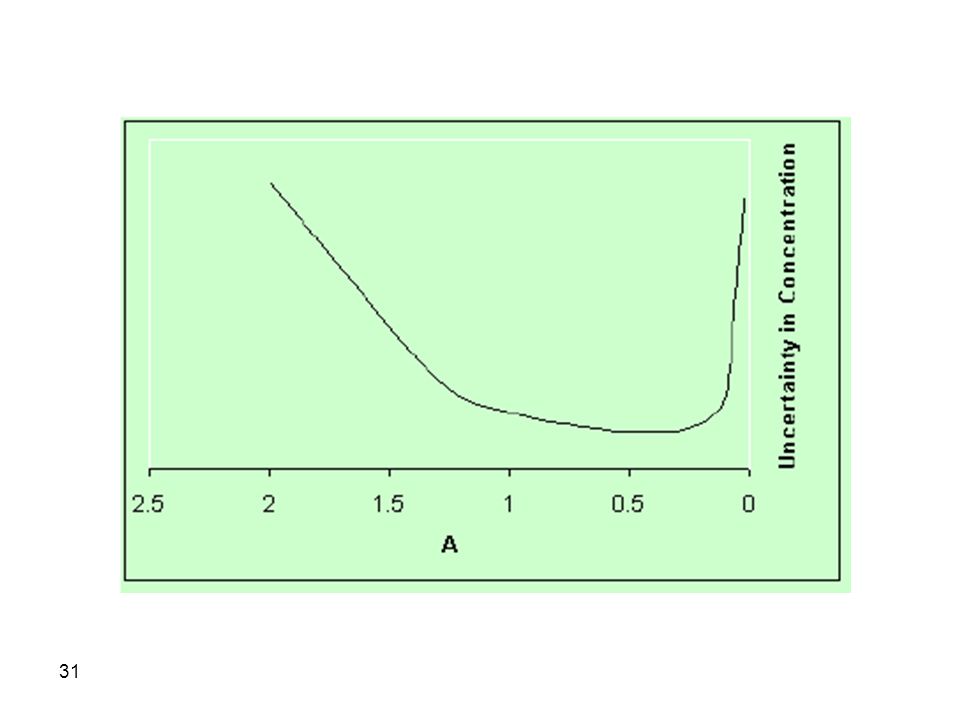
Therefore, an absorbance between 0. 2-0
Therefore, an absorbance between may be advantageous in terms of a lower uncertainty in concentration measurements. At higher or lower absorbances, an increase in uncertainty is encountered. It is therefore advised that the test solution be in the concentration range which gives an absorbance value in the range from for best precision. However, it should also be remembered that we ended up with this conclusion provided that sT is constant. Unfortunately, sT is not always constant which complicates the conclusions above.

33
EFFECT OF bandwidth WIDTH
Effect of bandwidth on spectral detail for a sample of benzene vapor. Note that as the spectral bandwidth increases, the fine structure in the spectrum is lost. At a bandwidth of 10 nm, only a broad absorption band is observed.

34
Effect of slit width (spectral bandwidth) on peak heights
Effect of slit width (spectral bandwidth) on peak heights. Here, the sample was s solution of praseodymium chloride. Note that as the spectral bandwidth decreases by decreasing the slit width from 1.0 mm to 0.1 mm, the peak heights increase.
 on peak heights. Here, the sample was s solution of praseodymium chloride. Note that as the spectral bandwidth decreases by decreasing the slit width from 1.0 mm to 0.1 mm, the peak heights increase.")
35
Effect of Scattered Radiation at Wavelength Extremes of an Instrument
Wavelength extremes of an instrument are dependent on type of source, detector and optical components used in the manufacture of the instrument. Outside the working range of the instrument, it is not possible to use it for accurate determinations. However, the extremes of the instrument are very close to the region of invalid instrumental performance and would thus be not very accurate. An example may be a visible photometer which, in principle, can be used in the range from nm. It may be obvious that glass windows, cells and prism will start to absorb significantly below 380 nm and thus a decrease in the incident radiant power is significant.

36
B: UV-VIS spectrophotometer
A: VIS spectrophotometer EFFECT OF SCATTERED RADIATION Spectrum of cerium (IV) obtained with a spectrophotometer having glass optics (A) and quartz optics (B). The false peak in A arises from transmission of stray radiation of longer wavelengths.
 obtained with a spectrophotometer having glass optics (A) and quartz optics (B). The false peak in A arises from transmission of stray radiation of longer wavelengths.")
37
The output from the source at the low wavelength range is minimal
The output from the source at the low wavelength range is minimal. Also, the detector has best sensitivities around 550 nm which means that away up and down this value, the sensitivity significantly decrease. However, scattered radiation, and stray radiation in general, will reach the detector without passing through these surfaces as well as these radiation are constituted from wavelengths for which the detector is highly sensitive. In some cases, stray and scattered radiation reaching the detector can be far more intense than the monochromatic beam from the source. False peaks may appear in such cases and one should be aware of this cause of such peaks.

38
Light Sources (commercial instruments) D2 lamp (UV: 160 – 375 nm)
Instrumentation Light source - selection Sample container Detector Signal processing Light Sources (commercial instruments) D2 lamp (UV: 160 – 375 nm) W lamp (vis: 350 – 2500 nm)
 D2 lamp (UV: 160 – 375 nm) W lamp (vis: 350 – 2500 nm)")
39
Sources Deuterium and hydrogen lamps (160 – 375 nm)
D2 + Ee → D2* → D’ + D’’ + h Excited deuterium molecule with fixed quantized energy Dissociated into two deuterium atoms with different kinetic energies Ee = ED2* = ED’ + ED’’ + hv Ee is the electrical energy absorbed by the molecule. ED2* is the fixed quantized energy of D2*, ED’ and ED’’ are kinetic energy of the two deuterium atoms.

41
Sources Deuterium lamp UV region
(a) A deuterium lamp of the type used in spectrophotometers and (b) its spectrum. The plot is of irradiance Eλ (proportional to radiant power) versus wavelength. Note that the maximum intensity occurs at ~225 m.Typically, instruments switch from deuterium to tungsten at ~350 nm.
 A deuterium lamp of the type used in spectrophotometers and (b) its spectrum. The plot is of irradiance Eλ (proportional to radiant power) versus. wavelength. Note that the maximum intensity occurs at ~225 m.Typically, instruments switch from deuterium to tungsten at ~350 nm.")
42
(a) A tungsten lamp of the type used in spectroscopy
Visible and near-IR region (a) A tungsten lamp of the type used in spectroscopy and its spectrum (b). Intensity of the tungsten source is usually quite low at wavelengths shorter than about 350 nm. Note that the intensity reaches a maximum in the near-IR region of the spectrum (~1200 nm in this case).
 A tungsten lamp of the. type used in spectroscopy. and its spectrum (b). Intensity of the tungsten. source is usually quite low. at wavelengths shorter than about 350 nm. Note that the intensity reaches a maximum in the near-IR. region of the spectrum. (~1200 nm in this case).")
43
The tungsten lamp is by far the most common source in the visible and near IR region with a continuum output wavelength in the range from nm. The lamp is formed from a tungsten filament heated to about 3000 oC housed in a glass envelope. The output of the lamp approaches a black body radiation where it is observed that the energy of a tungsten lamp varies as the fourth power of the operating voltage.

44
Tungsten halogen lamps are currently more popular than just tungsten lamps since they have longer lifetime. Tungsten halogen lamps contain small quantities of iodine in a quartz envelope. The quartz envelope is necessary due to the higher temperature of the tungsten halogen lamps (3500 oC). The longer lifetime of tungsten halogen lamps stems from the fact that sublimed tungsten forms volatile WI2 which redeposits on the filament thus increasing its lifetime. The output of tungsten halogen lamps are more efficient and extend well into the UV.
. The longer lifetime of tungsten halogen lamps stems from the fact that sublimed tungsten forms volatile WI2 which redeposits on the filament thus increasing its lifetime. The output of tungsten halogen lamps are more efficient and extend well into the UV..")
45
Sources Tungsten lamps (350-2500 nm)
Why add I2 in the lamps? W + I2 → WI2 Low limit: 350 nm Low intensity Glass envelope

46
3. Xenon Arc Lamps Passage of current through an atmosphere of high pressured xenon excites xenon and produces a continuum in the range from nm with maximum output at about 500 nm. Although the output of the xenon arc lamp covers the whole UV and visible regions, it is seldom used as a conventional source in the UV-Vis. The radiant power of the lamp is very high as to preclude the use of the lamp in UV-Vis instruments. However, an important application of this source will be discussed in luminescence spectroscopy which will be discussed later

48
Sample Containers Sample containers are called cells or cuvettes and are made of either glass or quartz depending on the region of the electromagnetic spectrum. The path length of the cell varies between 0.1 and 10 cm but the most common path length is 1.0 cm. Rectangular cells or cylindrical cells are routinely used. In addition, disposable polypropylene cells are used in the visible region. The quality of the absorbance signal is dependent on the quality of the cells used in terms of matching, cleaning as well as freedom from scratches.

49
Instrumental Components
Source - selection (monochromators) Sample holders Cuvettes (b = 1 cm typically) Glass (Vis) Fused silica (UV+Vis) Detectors Photodiodes PMTs
 Sample holders. Cuvettes (b = 1 cm typically) Glass (Vis) Fused silica (UV+Vis) Detectors. Photodiodes. PMTs.")
50
1. Single beam Place cuvette with blank (i.e., solvent) in instrument and take a reading 100% T Replace cuvette with sample and take reading % T for analyte (from which absorbance is calc’d)
")
51
Instrumentation Most common spectrophotometer: Spectronic 20.
On/Off switch and zero transmission adjustment knob Wavelength selector/Readout Sample chamber Blank adjustment knob Absorbance/Transmittance scale

53
End view of the exit slit of the Spectronic 20
spectrophotometer pictured earlier

54
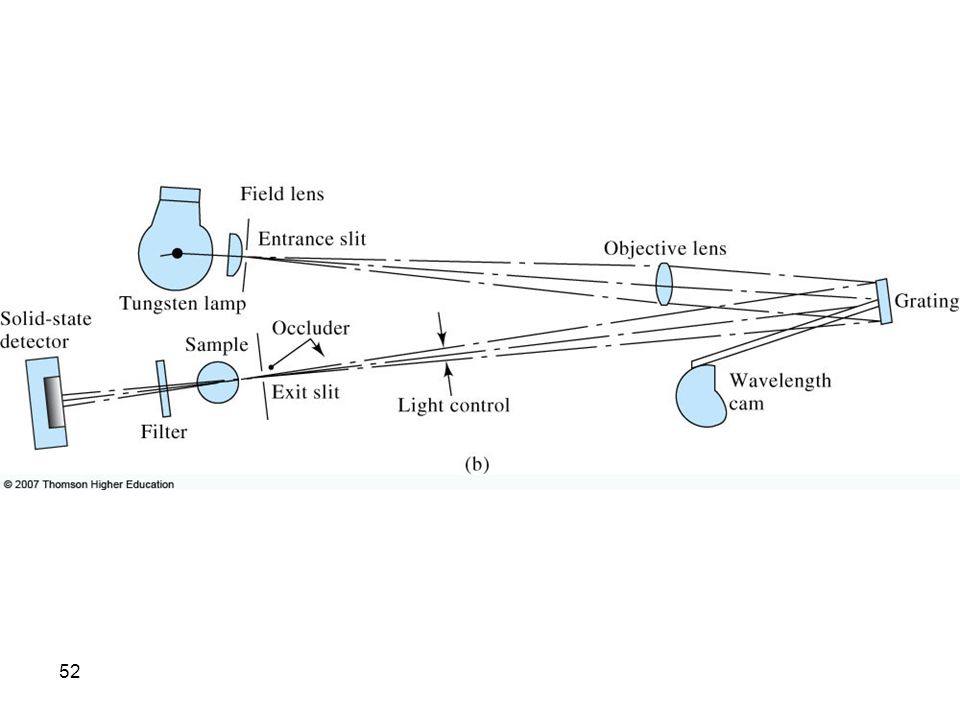
Single-Beam Instruments for the Ultraviolet/Visible Region

55
Single-Beam Computerized Spectrophotometers
Inside of a single-beam spectrophotometer connected to a computer.

56
Types of Instruments Instrumental designs for UV-visible photometers or spectrophotometers. In (a), a single-beam instrument is shown. Radiation from the filter or monochromator passes through either the reference cell or the sample cell before striking the photodetector.
, a single-beam instrument is shown. Radiation from the filter or monochromator passes through either the reference cell or the sample cell before striking the photodetector.")
58
2. Double beam (most commercial instruments)
Light is split and directed towards both reference cell (blank) and sample cell Two detectors; electronics measure ratio (i.e., measure/calculate absorbance) Advantages: Compensates for fluctuations in source intensity and drift in detector Better design for continuous recording of spectra
 and sample cell. Two detectors; electronics measure ratio (i.e., measure/calculate absorbance) Advantages: Compensates for fluctuations in source intensity and drift in detector. Better design for continuous recording of spectra.")
59
General Instrument Designs
Double Beam: In - Space Needs two detectors 59

60
General Instrument Designs
Double Beam: In - Time

62
Merits of Double Beam Instruments
Compensate for all but the most short term fluctuation in radiant output of the source Compensate drift in transducer and amplifier Compensate for wide variations in source intensity with wavelength

63
Location of Sample cell
In all photometers and scanning spectrophotpmeters described above, the cell has been positioned after the monochromators. This is important to decrease the possibility of sample photodecomposition due to prolonged exposure to all frequencies coming from the source. However, the sample is positioned before the monochromator in multichannel instruments like a photodiode array spectrophotometer. This can be done without fear of photodecomposition since the sample exposure time is usually less than 1 s. Therefore, it is now clear that in UV-Vis where photodecomposition of samples can take place, the sample is placed after the monochromators in scanning instruments while positioning of the sample before the monochromators is advised in multichannel instruments.

64
3. Multichannel Instruments
Photodiode array detectors used (multichannel detector, can measure all wavelengths dispersed by grating simultaneously). Advantage: scan spectrum very quickly “snapshot” < 1 sec. Powerful tool for studies of transient intermediates in moderately fast reactions. Useful for kinetic studies. Useful for qualitative and quantitative determination of the components exiting from a liquid chromatographic column.
. Advantage: scan spectrum very quickly snapshot < 1 sec. Powerful tool for studies of transient intermediates in moderately fast reactions. Useful for kinetic studies. Useful for qualitative and quantitative determination of the components exiting from a liquid chromatographic column.")
65
Multi-channel Design

67
A multichannel diode-array spectrophotometer, the Agilent Technologies 8453.

68
4. Probe Type Instruments
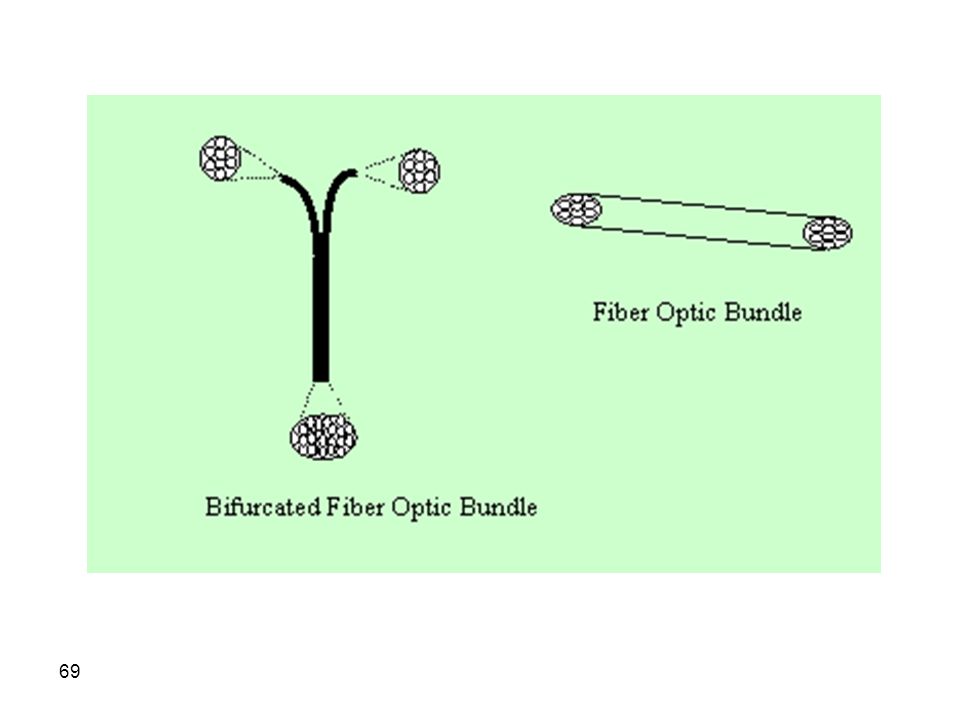
These are the same as conventional single beam instruments but the beam from the monochromators is guided through a bifurcated optical fiber to the sample container where absorption takes place. The attenuation in reflected beam at the specified wavelength is thus measured and related to concentration of analyte in the sample. A fiber optic cable can be referred to as a light pipe where light can be transmitted by the fiber without loss in intensity (when light hits the internal surface of the fiber at an angle larger than a critical angle). Therefore, fiber optics can be used to transmit light for very long distances without losses. A group of fibers can be combined together to form a fiber optic cable or bundle. A bifurcated fiber optic cable has three terminals where fibers from two separate cables are combined at one end to form the new configuration.
. Therefore, fiber optics can be used to transmit light for very long distances without losses. A group of fibers can be combined together to form a fiber optic cable or bundle. A bifurcated fiber optic cable has three terminals where fibers from two separate cables are combined at one end to form the new configuration.")
70
Fiber optic probe

72
Double Dispersing Instruments
The instrument in this case has two gratings where the light beam leaving the first monochromators at a specified wavelength is directed to the second grating. This procedure results in better spectral resolution as well as decreased scattered radiation. However, double dispersing instruments are expensive and seem to offer limited advantages as compared to cost; especially in the UV-Vis region where exact wavelength may not be crucial.

73
Optical diagram of the Varian Cary 300 double-dispersing spectrophotometer. A second monochromator is added immediately after the source.

75
Molar absorptivities e = 8.7 x 10 19 P A
A: cross section of molecule in cm2 (~10-15) P: Probability of the electronic transition (0-1) P>0.1-1 allowable transitions P<0.01 forbidden transitions
 P: Probability of the electronic transition (0-1) P>0.1-1 allowable transitions. P<0.01 forbidden transitions.")
76
Molecular Absorption M + hn M* (absorption 10-8 sec)
M* M + heat (relaxation process) M* A+B+C (photochemical decomposition) M* M + hn (emission)
 M* A+B+C (photochemical decomposition) M* M + hn (emission)")
77
Visible Absorption Spectra

78
The absorption of UV-visible radiation generally results from excitation of bonding electrons.
can be used for quantitative and qualitative analysis

79
Molecular orbital is the nonlocalized fields between atoms that are occupied by bonding electrons. (when two atom orbitals combine, either a low-energy bonding molecular orbital or a high energy antibonding molecular orbital results.) Sigma () orbital The molecular orbital associated with single bonds in organic compounds Pi () orbital The molecular orbital associated with parallel overlap of atomic P orbital. n electrons No bonding electrons
 orbital. The molecular orbital associated with single bonds in organic compounds. Pi () orbital. The molecular orbital associated with parallel overlap of atomic P orbital. n electrons. No bonding electrons.")
80
Molecular Transitions for UV-Visible Absorptions
What electrons can we use for these transitions?

81
MO Diagram for Formaldehyde (CH2O)
s = p = n =

82
Singlet vs. triplet In these diagrams, one electron has been excited (promoted) from the n to * energy levels (non-bonding to anti-bonding). One is a Singlet excited state, the other is a Triplet.

83
Type of Transitions σ → σ* n → σ* n → * and → *
High energy required, vacuum UV range CH4: = 125 nm n → σ* Saturated compounds, CH3OH etc ( = nm) n → * and → * Mostly used! = nm
 n → * and → * Mostly used! = nm.")
84
UV-Visible Absorptions
Examples of UV-Visible Absorptions LOW!

85
UV-Visible Absorption Chromophores

86
Effects of solvents Blue shift (n- p*) (Hypsocromic shift)
Increasing polarity of solvent better solvation of electron pairs (n level has lower E) peak shifts to the blue (more energetic) 30 nm (hydrogen bond energy) Red shift (n- p* and p –p*) (Bathochromic shift) Increasing polarity of solvent, then increase the attractive polarization forces between solvent and absorber, thus decreases the energy of the unexcited and excited states with the later greater peaks shift to the red 5 nm
 peak shifts to the blue (more energetic) 30 nm (hydrogen bond energy) Red shift (n- p* and p –p*) (Bathochromic shift) Increasing polarity of solvent, then increase the attractive polarization forces between solvent and absorber, thus decreases the energy of the unexcited and excited states with the later greater. peaks shift to the red. 5 nm.")
87
UV-Visible Absorption Chromophores

88
Typical UV Absorption Spectra
Chromophores?

89
The effects of substitution
Auxochrome function group Auxochrome is a functional group that does not absorb in UV region but has the effect of shifting chromophore peaks to longer wavelength as well As increasing their intensity.

90
Now solvents are your “container”
They need to be transparent and do not erase the fine structure arising from the vibrational effects Polar solvents generally tend to cause this problem Same solvent must be Used when comparing absorption spectra for identification purpose.

92
Summary of transitions for organic molecules
s s* transition in vacuum UV (single bonds) n s* saturated compounds with non-bonding electrons l ~ nm e ~ ( not strong) n p*, p p* requires unsaturated functional groups (eq. double bonds) most commonly used, energy good range for UV/Vis l ~ nm n p* : e ~ p p*: e ~ 1000 – 10,000
 n s* saturated compounds with non-bonding electrons. l ~ nm. e ~ ( not strong) n p*, p p* requires unsaturated functional groups (eq. double bonds) most commonly used, energy good range for UV/Vis. l ~ nm. n p* : e ~ p p*: e ~ 1000 – 10,000.")
93
List of common chromophores and their transitions

94
Organic Compounds Most organic spectra are complex
Electronic and vibration transitions superimposed Absorption bands usually broad Detailed theoretical analysis not possible, but semi-quantitative or qualitative analysis of types of bonds is possible. Effects of solvent & molecular details complicate comparison

95
Rule of thumb for conjugation
If greater then one single bond apart - e are relatively additive (hyperchromic shift) - l constant CH3CH2CH2CH=CH2 lmax= emax = ~10,000 CH2=CHCH2CH2CH=CH2 lmax= emax = ~20,000 If conjugated - shifts to higher l’s (red shift) H2C=CHCH=CH2 lmax=217 emax = ~21,000
 - l constant. CH3CH2CH2CH=CH2 lmax= 184 emax = ~10,000. CH2=CHCH2CH2CH=CH2 lmax=185 emax = ~20,000. If conjugated. - shifts to higher l’s (red shift) H2C=CHCH=CH2 lmax=217 emax = ~21,000.")
96
Spectral nomenclature of shifts

97
What about inorganics? Common anions np* nitrate (313 nm), carbonate (217 nm) Most transition-metal ions absorb in the UV/Vis region. In the lanthanide and actinide series the absorption process results from electronic transitions of 4f and 5f electrons. For the first and second transition metal series the absorption process results from transitions of 3d and 4d electrons. The bands are often broad. The position of the maxima are strongly influenced by the chemical environment. The metal forms a complex with other stuff, called ligands. The presence of the ligands splits the d-orbital energies.

98
Transition metal ions

99
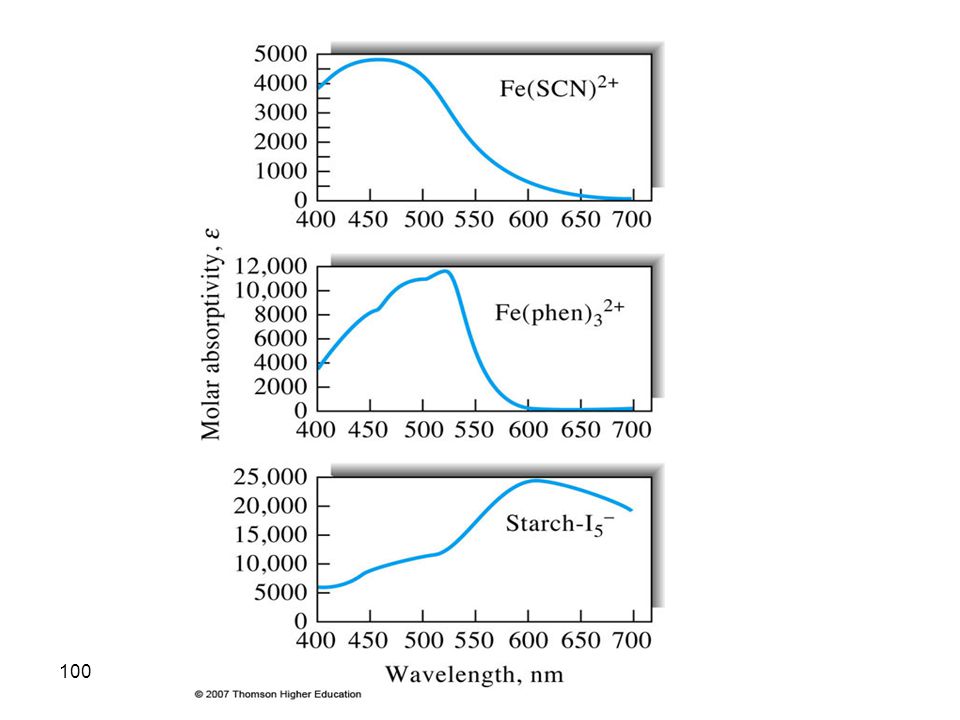
Charge-Transfer-Absorption
A charge-transfer complex consists of an electron-donor group bonded to an electron acceptor. When this product absorbs radiation, an electron from the donor is transferred to an orbital that is largely associated with the acceptor. Large molar absorptivity (εmax >10,000) Many organic and inorganic complexes
 Many organic and inorganic complexes.")
Similar presentations