Download presentation
Presentation is loading. Please wait.

1
Inborn errors of Metabolism
Approach to Diagnosis and management

2
Defining inborn errors of metabolism
Broad definition: Any defect related to the inappropriate metabolism of substrates essential for an organism’s survival Defects in energy metabolism Defects in protein synthesis or catabolism leading to intoxication with toxic substrates and/or absence of essential metabolic substrates Synthetic defects – inability to produce an essential structural substrate Defects in transport of essential components – cystic fibrosis, lysinuric protein deficiency Defects in detoxifying specific substances Generalized global tissue dysfunction (mitochondria)
")
3
Inborn errors of metabolism: General points
Incidence in heterogeneous population 1: However, incidence higher if we include hyperlipoproteinemias Heterozygote familial hypercholesterolemia has an incidence of 1:500 Much more common in inbred populations and consanguineous couples Signs and symptoms of metabolic disease are usually non-specific Can present at all ages Underdiagnosis of metabolic diseases but improving with new genetic techniques and improved mass screening

4
Mortality related to metabolic diseases
120 children aged 0-14 y died in a ten year period of inborn errors of metabolism This was 3.5% of total child mortality and 9.5% of non-acquired (genetic) mortality
 mortality.")
5
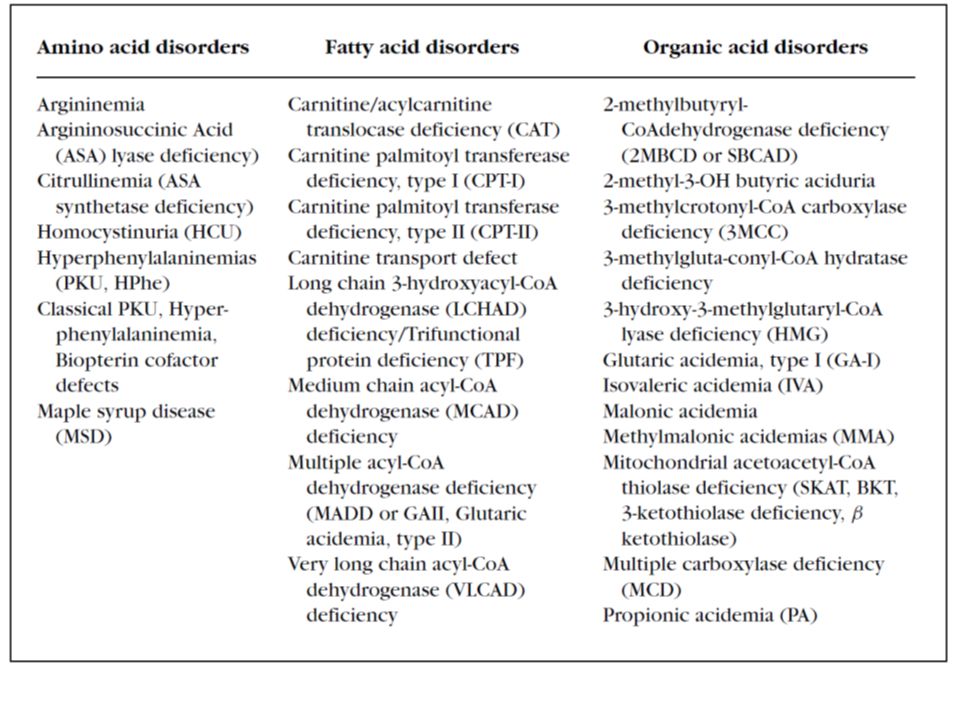
Complexity of inborn errors of metabolism

7
Diagnosing metabolic disease

8
Approach to diagnosing metabolic disease
Genetic testing for specific at risk groups Prevention Screening approach Testing entire population using relatively inexpensive (price/unit) techniques High throughput (180,000 samples/yr/IL) Clinical, biochemical, and molecular analysis of symptomatic patients
 techniques. High throughput (180,000 samples/yr/IL) Clinical, biochemical, and molecular analysis of symptomatic patients.")
9
Populations at risk Specific population groups are at greater risk for a disease Founder effect Inbreeding Prevention Premarital testing Prenatal testing Preimplantation genetic diagnosis

10
Basis for newborn screening
Cost-effective Easy/inexpensive method for detecting IEM High throughput methods Rapid turnaround High sensitivity (detect most cases) High specificity (low false positive rate) Effective notification system Possibility for intervention Minimal harm to patient
 High specificity (low false positive rate) Effective notification system. Possibility for intervention. Minimal harm to patient.")
11
Newborn screening First introduced for PKU (Guthrie) Ames test
Extended to several other diseases including galactosemia, hypothyroidism, sickle cell, thallasemia, congenital adrenal hyperplasia, and homocystinuria More countries and states including Israel are now using tandem mass spectrometry for more extensive evaluation

12
WHO criteria for screening

16
Early methods for screening for inborn errors of metabolism
Ames test Paper impregnated with ferric chloride which reacts with phenylalanine Guthrie test Place blood drop on agar plate containing B. subtilis and an inhibitor B-2-thienylalanine High levels of phenylalanine in blood spot overcome inhibition and there is growth

17
Tandem mass spectrometry (MS/MS)
")
20
Approximately .3/1000 detection rate

21
Phenylketonuria Phenylalanine hydroxylase

22
Phenylketonuria In 1934, Folling discovered that children with mental retardation had high levels of phenylalanine in blood Classic: deficiency in phenylalanine hydroxylase Abnormalities in tone, myoclonic seizures, mental retardation, disorders of pigmentation Incidence: 1;10,000-15,000, Turkey: <1:3000 TX: Dietary – reduction in phenylalanine, provision of tyrosine

23
CONTROL PKU MSUD

24
Problems with newborn screening
Not all diseases are screened False positive – without disease False negative – disease undetected Some disorders are not disorders Asymptomatic children Unclear treatment recommendations Difficulty in follow-up Privacy issues

25
Disorders not detected
Disorders of carbohydrate metabolism Mitochondrial disorders Steroid disorders and hyperlipidemias Some urea cycle disorders Tissue specific disorders with no blood markers

26
CASE STUDY: Short chain acyl dehydrogenase (SCAD) deficiency
Associated with CNS disease Question of whether this gene should be screened for US vs. Europe High prevalence of 319 C>T mutation among Ashkenazic Jews 1:15 carrier rate, 1:900 homozygotes Should be 2700 cases in Israel but only a handful of symptomatic individuals What type of intervention?

32
Tay-Sachs Hexoaminidase A deficiency Cerebral and retinal degeneration
Cherry red spot (macular) Hypotonia, motor weakness, developmental delay Carrier rate 1:30 in Ashkenazic Jews Screening has led to 90% reduction in Jewish population
 Hypotonia, motor weakness, developmental delay. Carrier rate 1:30 in Ashkenazic Jews. Screening has led to 90% reduction in Jewish population.")
33
Signs of metabolic disease
If the child: Looks bad Smells bad Feels bad Tastes bad Sounds bad Think Metabolic

34
Manifestations of metabolic diseases
CNS Neuroanatomic malformations Leigh’s disease Developmental delay, mental retardations Movement disorders Hypotonia, spasticity Altered mental status Seizures Visual disturbances Pigmented retinopathy Deafness

35
Manifestations of metabolic diseases
Cardiac Cardiomegaly (Pompe’s disease) Dilated cardiomyopathy Rhythm Disturbances Pulmonary Proteinosis (lysinuric protein intolerance) Insufficiency Frequent pneumonia Tachypnea, Kussmaul breathing (acidosis)
 Dilated cardiomyopathy. Rhythm Disturbances. Pulmonary. Proteinosis (lysinuric protein intolerance) Insufficiency. Frequent pneumonia. Tachypnea, Kussmaul breathing (acidosis)")
36
Manifestations of metabolic diseases
GI Dysphagia Pseudo-obstruction, diarrhea Bloating Liver Insufficiency Inflammation (increased transaminases) Hyperbilirubinemia Cirrhosis Hepatomegaly (glycogen or fats)
 Hyperbilirubinemia. Cirrhosis. Hepatomegaly (glycogen or fats)")
37
Manifestations of metabolic diseases
Renal Insufficiency Storage disease RTA Musculoskeletal Malformations Osteopenia/osteoporosis Weakness Myoglobinuria Contractures

38
Manifestations of metabolic diseases
Skin Nearly all skin disorders including seborrhea, ichthyosis Hematological Anemia (all types) Thrombocytopenia/Leukopenia Pancytopenia
 Thrombocytopenia/Leukopenia. Pancytopenia.")
39
Manifestations of mitochondrial disease
Cardiomyopathy / myopathy Stroke-like episodes, neuroanatomic changes Lactic acidosis, Leigh syndrome Seizures, myoclonues, dystonia, Parkinsonism Diabetes Deafness Chorea, ataxia, encephalomyelopathy Sudden infant death syndrome Ophalmoplegia, optic neuropathy, pigmented retinopathy Nelson, Textbook of Pediatrics, 19th edition, p. 508

40
Manifestations of metabolic diseases
Biochemical Hypoglycemia Endocrine disturbances Metabolic acidosis Respiratory acidosis/alkalosis Lactic acidosis Hyperammonemia Hypoalbuminemia Altered coagulation profile Hepatitis profile

41
Evaluation of infants with suspected metabolic disorders
Primary evaluation Arterial blood gas Glucose CBC and Differential Lactate/pyruvate Urinary reducing substances Serum ammonia Liver function tests PT/PTT NOTE: Very important at this step to rule out other non-metabolic related causes for clinical manifestations

42
Evaluation of infants with suspected metabolic disorders
Secondary evaluation Urine organic acids Serum amino acids Total and free plasma carnitine Very long chain fatty acids Fed and fasting lactate and pyruvate Cardiac echo Opthalmologic exam EEG Head CT and/or MRI

43
Evaluation of infants with suspected metabolic disorders
Tertiary evaluation Depends on results of initial evaluation Specific enzyme assays in lymphocytes, fibroblasts or tissue Specialized tests, e.g., CSF glycine or lactate, acylcarnitine profile, glycoprotein electrophoresis Histology, e.g., muscle biopsy, histological staining Mitochondrial studies Activity O2 Uptake DNA analysis Challenge tests – fasting, protein loading

44
Genetic analysis Mutational analysis Reverse genetics
When specific defect is known Reverse genetics Arrays, chips, genomic scanning, SNPs, Cost effective Excellent coverage Does not always identify correct genetic defect or sole defect Advantages over the traditional approach

45
Treatment of metabolic diseases
Enzyme replacement Detoxification Urea cycle defects Nutritional support Bypass defect NTBC in tyrosinemia Cofactor treatment Transplantation Genetic therapy Bone marrow Stem cell

46
Galactosemia Classic disease caused by galactose-1-phosphate uridyl transferase Symptoms after lactose formula or breast feeding Hepatomegaly and jaundice Gram- sepsis Cataracts if untreated Tx: non-lactose containing formula

47
Tyrosinemia Type 1 Defect in Fumarylacetoacetate hydrolase
Hepatic decompensation, coagulopathy, low albumin, cirrhosis, RTA DX: elevated tyrosine, characteristic urine metabolites including succinylacetone Long term – hepatic carcinoma TX: Low tyrosine/phenylalanine diet, treatment with NTBC, a herbicide, which inhibits hydroxyphenyl-pyruvate hydroxylase

Similar presentations

















 Dr Mohammad Khassawneh Assistant professor of pediatrics.>")





 LIVER FUNCTION AND THE BILIARY TRACT LECTURE FOUR Dr. Essam H. Aljiffri.>")



.>")


