Download presentation
Presentation is loading. Please wait.

1
Introduction to Real-Time PCR
Dr. Chaim Wachtel April 11, 2013

2
Real-Time PCR What is it? How does it work
How do you properly perform an experiment Analysis

3
The Nobel Prize in Chemistry 1993 was awarded "for contributions to the developments of methods within DNA-based chemistry" jointly with one half to Kary B. Mullis "for his invention of the polymerase chain reaction (PCR) method"and with one half to Michael Smith "for his fundamental contributions to the establishment of oligonucleotide-based, site-directed mutagenesis and its development for protein studies". Michael Smith

4
PCR – A simple idea Polymerase Chain Reaction: Kary Mullis (1983)
In vitro method for enzymatically synthesizing DNA The reaction uses two oligonucleotide primers that hybridize to opposite strands and flank the target DNA sequence that is to be amplified A repetitive series of cycles gives exponential accumulation of a specific DNA fragment Template denaturation Primer annealing Extension of annealed primers by the polymerase The number of target DNA copies doubles every PCR cycle (20 cycles 220≈106 copies of target)
")
5
Principle of PCR

6
Difference PCR vs real-time PCR?
Fluorescence is measured every cycle (signal amount of PCR product). Curves rise after a number of cycles that is proportional to the initial amount of DNA template. Comparison with standard curve gives quantification.
. Curves rise after a number of cycles that is proportional to the initial amount of DNA template. Comparison with standard curve gives quantification.")
7
Real-Time and End Point

8
MIQE: the minimum information for the publication of qPCR experiments.

12
The mRNA of the Arabidopsis Gene FT Moves from Leaf to Shoot Apex and Induces Flowering
Tao Huang, Henrik Böhlenius, Sven Eriksson, François Parcy, and Ove Nilsson Science 9 September 2005: 2005: Signaling Breakthroughs of the Year

13
Retraction WE WISH TO RETRACT OUR RESEARCH ARTICLE “THE MRNA OF THE ARABIDOPSIS GENE FT MOVES from leaf to shoot apex and induces flowering” (1). After the first author (T.H.) left the Umeå Plant Science Centre for another position, analysis of his original data revealed several anomalies. It is apparent from these files that data from the real-time RT-PCR were analyzed incorrectly. Certain data points were removed, while other data points were given increased weight in the statistical analysis. When all the primary real-time RT-PCR data are subjected to correct statistical analysis, most of the reported significant differences between time points disappear. Because of this, we are retracting the paper in its entirety.
. After the first author (T.H.) left the Umeå Plant Science Centre for another position, analysis of his original data revealed several anomalies. It is apparent from these files that data from the real-time RT-PCR were analyzed incorrectly. Certain data points were removed, while other data points were given increased weight in. the statistical analysis. When all the primary real-time RT-PCR data are subjected to correct statistical analysis, most of the reported significant differences between time points disappear. Because of this, we are retracting the paper in its entirety.")
14
Real-Time Machines How do they work What can you do with one
Gene expression SNP detection DNA detection (quantify) How do you use them Experiment design Everything you need to know and more about RNA and RT-PCR
 How do you use them. Experiment design. Everything you need to know and more about RNA and RT-PCR.")
15
First real-time PCR, 1991 PCR tube in thermocycler
spectrofluorometer fiberoptic “Fifty Years of Molecular Diagnostics” Clin Chem Mar;51(3): (C.Wittwer, ed.)
: (C.Wittwer, ed.)")
16
First commercial real-time PCR instruments
ABI 7700 – laser/fiberoptic-based ABI 5700 – CCD camera-based Idaho Technology LightCycler – capillary tubes

17
RT-PCR machines at Bar Ilan
7900HT Fast Real-Time PCR System (Sol Efroni’s lab) AB StepOnePlus Fast Real-Time PCR System Qiagen’s Rotor-gene (Oren Levy’s lab) Thermo PikoReal (Bachelet Lab) Bio-Rad CFX-96
 AB StepOnePlus. Fast Real-Time PCR System. Qiagen’s Rotor-gene. (Oren Levy’s lab) Thermo PikoReal. (Bachelet Lab) Bio-Rad CFX-96.")
18
Rotor-gene

19
Probing alternatives Non-specific detection
Dyes: SYBR Green I, BEBO, BOXTO, EvaGreen... Specific detection TaqMan probe Molecular Beacon Light-Up probe Hybridization probes Primer based detection Scorpion primers QZyme Lux primers

20
SYBR Green binds to dsDNA
SYBR Green binds to DNA, particularly to double-stranded DNA, giving strongly enhanced fluorescence. SYBR Green is sequence-dependent!

21
The TaqMan Probe The TaqMan probe binds to ssDNA at a combined annealing and elongation step. It is degraded by the polymerase, which releases the dye from the quencher.

22
Multiplex Q-PCR Detection of two (or more) different target sequences in the same reaction.
 different target sequences in the same reaction.")
23
qPCR technical workflow
DNA Extraction Data Analysis Sampling qPCR RNA Extraction DNase treatment Reverse Transcription

24
Nucleic acid isolation and purification
24

25
Overview Sampling Accessibility and lysis Commonly used techniques
RNA considerations Quality control The process of purifying nucleic acids is often taken for granted Researchers frequently underestimate the importance of choosing a method that gives them not only good recoveries but also molecular biology-grade samples A lot of time and money are invested in molecular biology applications, so its important to isolate highly pure nucleic acids to begin with Without an effective method for nucleic acid purification, the quality of the results will be compromised. 25

26
Why sample preparation?
Make target available Remove inhibitors Remove fluorescent contaminants Preserve target integrity Concentrate target

27
Path Reverse Transcription Real-time PCR DNA RNA Purification
Isolation Disruption mRNA Genomic DNA Total RNA Plasmid DNA Fragment DNA Nuclear RNA Phage DNA

28
Accessibility Sample disruption and homogenization Mechanical
Grinding, Sonication, Vortexing, Polytron Physical Freezing Enzymatic Proteinase K, Lysozyme, Collagenase Chemical Guanidinium isothiocyanate (GITC), Alkali treatment, CTAB Disruption is the physical breakdown of cellular components (walls and plasma membranes) using mechanical and/or enzymatic techniques. Successful disruption will release all RNA contained in the sample and produce high yield Homogenization will reduce the viscosity of the cell lysate and incorporate large particles formed in the disruption step into a homogenous mixture. Successful homogenization will reduce column clogging and generate high yield Nämn även beta-mercaptoetanol hexadecyltrimethylammonium bromide (CTAB) Mechanical methods for disrupting fresh tissue and cells include homogenization with a Dounce or with a mechanical homogenizer (such as the Brinkmann Polytron¥), vortexing, sonication, French press, bead milling, and even grinding in a coffee grinder! 28
, Alkali treatment, CTAB. Disruption is the physical breakdown of cellular components (walls and plasma membranes) using mechanical and/or enzymatic techniques. Successful disruption will release all RNA contained in the sample and produce high yield. Homogenization will reduce the viscosity of the cell lysate and incorporate large particles formed in the disruption step into a homogenous mixture. Successful homogenization will reduce column clogging and generate high yield. Nämn även beta-mercaptoetanol. hexadecyltrimethylammonium bromide (CTAB) Mechanical methods for disrupting fresh tissue and cells include homogenization with a Dounce or with a mechanical homogenizer (such as the Brinkmann Polytron¥), vortexing, sonication, French press, bead milling, and even grinding in a coffee grinder! 28.")
29
Lysis Complete or partial lysis? Chaotropic lysis buffers:
SDS, GITC, LiCl, phenol, sarcosyl Gentle lysis buffers: NP-40, Triton X-100, Tween, DTT More strong: urea, chloroform, They are chaotropic detergents Weak lysis could used to du plasma membrane specific lysis NP-40 = Igepal CA-630 Sarcosyl is also called N-laurylsarcosine and is mainly used for plant samples 29

30
Purification principles
Characteristics of nucleic acids Long, unbranched, negatively charged polymers Examples: Differential solubility Precipitation Strong affinity to surface Factors: pH, [salt], hydrophobicity

31
Purification techniques
Solution based- eg Tri reagent, CsCl gradient Precipitation- ethanol, needs salt, multiple factors can influence precipitation Membrane based- spin columns (Qiagen and the like) Magnetic bead based Typical protocol: Stabilisation of the sample Lysis and homogenisation in denaturing buffer / beta-mercaptoethanol Addition of ethanol for adjustment of binding conditions Selective RNA binding (nature and concentration of the chaotrope) Stringent salt wash – removal of proteins, carbohydrates and most gDNA DNAse I digest (optional) Second stringent wash Ethanolic wash Dry spin / extended vacuum step Elution in TE buffer or RNase-free water 31
 Magnetic bead based. Typical protocol: Stabilisation of the sample. Lysis and homogenisation in denaturing buffer / beta-mercaptoethanol. Addition of ethanol for adjustment of binding conditions. Selective RNA binding (nature and concentration of the chaotrope) Stringent salt wash – removal of proteins, carbohydrates and most gDNA. DNAse I digest (optional) Second stringent wash. Ethanolic wash. Dry spin / extended vacuum step. Elution in TE buffer or RNase-free water. 31.")
32
Solution based isolation
Most methods use hazardous reagents Phenol/Chloroform extraction Proteins, lipids, polysaccharides go into the organic phase or in the interphase. DNA/RNA remains in aqueous phase Caesium chloride density gradient ultracentrifugation Time consuming Acid guanidine phenol chloroform extraction Commonly called TRIzol

33
Precipitation purification
Nucleic acids precipitate in alcohols Salt (NaCl, NaAc) facilitates the process Important factors: Temperature, time, pH, and amount
 facilitates the process. Important factors: Temperature, time, pH, and amount.")
34
Membrane based isolation
Anion exchange technology Spin column / silica gel membrane Chaotropic salts (e.g. NaI or guanidine hydrochloride) bind H2O molecules Loss of water from DNA changes shape and charge DNA binds reversibly to silica membrane
 bind H2O molecules. Loss of water from DNA changes shape and charge. DNA binds reversibly to silica membrane.")
35
Purification – GITC vs. column
Organic liquids Pro: Higher yield Can handle larger amounts of cells Better for troublesome tissues (fatty tissue, bone etc) Con: Higher DNA contamination (for RNA isolation) Separate DNase I digestion with additional purification Spin columns Pro: Less contaminating DNA (for RNA isolation) On column DNase digestion Less loss of RNA Higher quality Easy to use Con: Limited loading capacity More expensive (?)
 Con: Higher DNA contamination (for RNA isolation) Separate DNase I digestion with additional purification. Spin columns. Pro: Less contaminating DNA (for RNA isolation) On column DNase digestion Less loss of RNA. Higher quality. Easy to use. Con: Limited loading capacity. More expensive ( )")
36
RNA Considerations RNA is chemically and biologically less stable than DNA Extrinsic and intrinsic ribonucleases (RNases) Specific and Nonspecific inhibitors Speciellt känsligt är RNA vid >65 grader och i basisk miljö RNase finns överallt, även inne i alla celler Successful isolation depends on the inactivation of ribonucleases which degrade RNA rendering it useless for downstream analysis Reagents and plasticware should either be guaranteed RNase-free by the supplier or treated with DEPC Inactivated by the GITC and beta-mercaptoethanol present in the lysis buffer 36

37
Stabilizing conditions
Work on ice Process immediately Flash freeze sample in liquid nitrogen and store at -70°C until later use Store samples in stabilization buffer RNAlater: Permeates the sample and freezes the gene expression profile Entirely non-hazardous Can be used at ambient temperature Sample can be repeatedly frozen and thawed without reduction in RNA quality 37

38
Storage of nucleic acids
Nuclease-free plasticware Eluted in nuclease-free water, TE or sodium citrate solution RNA: Neutral pH to avoid degradation Aliquot sample to avoid multiple freeze-thaw cycles Isolated RNA should be stored at -20 deg C or -70 deg C for even better protection in ethanol and not water. milli Q / double distilled lab water is often at acid pH 38

39
Quality Control Spectroscopic methods
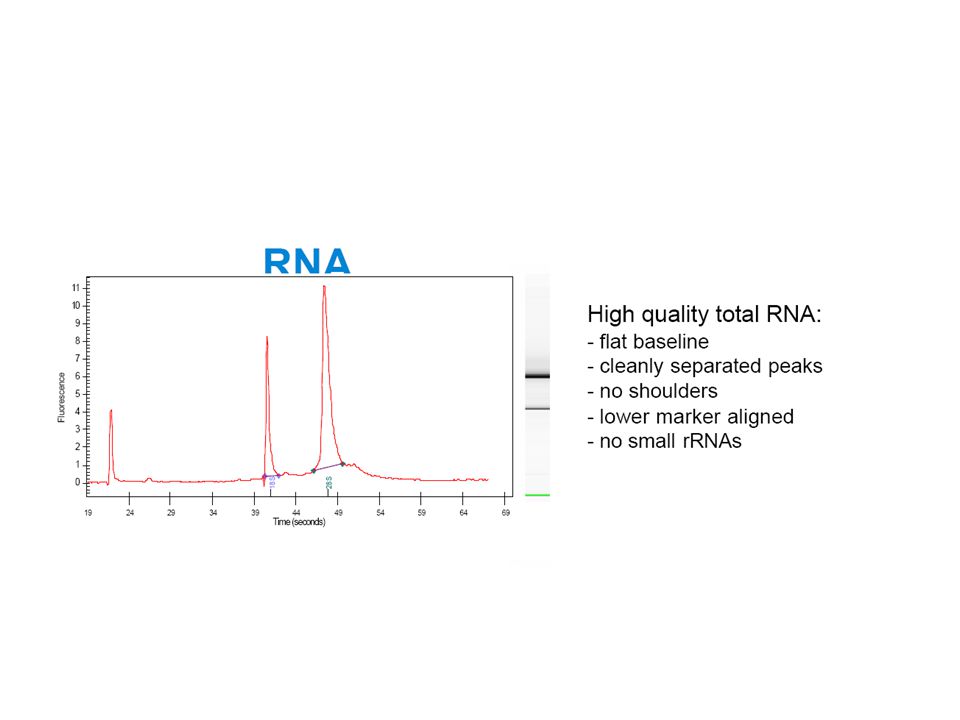
Concentration, [NA] = A260 x e mg/ml Purity: A260 / A280 (≈1.8 for DNA, 2.0 for RNA) Dyes Quantification by fluorescence of DNA/RNA-binding dyes (Qubit) Electrophoresis (28S and 18S bands) Kanke ändra bort epsilon till faktor. Extinction coefficient for DNA is 50, RNA it’s 40. Assuming l = 1 cm. Pure DNA has ratio of , pure RNA has Optimise the quantity of staring material Ensure reproducibility by always weighing the sample Thereby avoid exceeding the lysis capacity of the buffer Check lysis buffer for precipitation If following a vacuum protocol, then set the strength of the vacuum with empty wells If following a spin protocol, then use an appropriate g force at every spin step Determine the yield and quality spectrophotometrically or using the Agilent 2100 Bioanalyser 39
 Dyes. Quantification by fluorescence of DNA/RNA-binding dyes (Qubit) Electrophoresis (28S and 18S bands) Kanke ändra bort epsilon till faktor. Extinction coefficient for DNA is 50, RNA it’s 40. Assuming l = 1 cm. Pure DNA has ratio of , pure RNA has Optimise the quantity of staring material. Ensure reproducibility by always weighing the sample. Thereby avoid exceeding the lysis capacity of the buffer. Check lysis buffer for precipitation. If following a vacuum protocol, then set the strength of the vacuum with empty wells. If following a spin protocol, then use an appropriate g force at every spin step. Determine the yield and quality spectrophotometrically or using the Agilent 2100 Bioanalyser. 39.")
40
What is the BioAnalizer?
Microfluidic separations technology RNA - DNA - Protein 1µl of RNA sample (100 pg to 500 ng) 12 samples analyzed in 30 min Integrated analysis software: Quantitation Integrity of RNA
 12 samples analyzed in 30 min. Integrated analysis software: Quantitation. Integrity of RNA.")
41
Bioanalyzer

45
RNA Integrity: RQI Good RNA Quality Bad RNA Quality
10 RNA Quality Indicator 1

46
Publications on RNA integrity

47
DNase I treatment of RNA samples
RT, No DNase No RT, No DNase RT, DNase No RT, DNase

48
qPCR technical workflow
DNA Extraction Data Analysis Sampling qPCR RNA Extraction DNase treatment Reverse Transcription

49
Reverse transcription RT
49

50
Outline Priming efficiency Reproducibility
Properties of Reverse transcriptase RNA concentrations

51
General description of RT reaction
Reverse Transcriptases are RNA-dependent* DNA polymerases that catalyze first strand DNA synthesis in presence of a suitable primer+ as long as it has a free 3’ OH end. *Can use also single strand DNA as template. + Can be either RNA or DNA.

52
RT priming

53
RT with Gene-Specific Priming

54
RT with Oligo(dT) Priming
 Priming")
55
RT with Random Hexamer Priming

56
Real-time PCR using different RT primers

57
Real-time PCR with different RT primers

58
Dependence on priming strategy

59
Dependance of priming method
Gene b-tubulin CaVID GAPDH Insulin II Glut 2 hexamers 19,5 26,5 15,8 16,9 27,5 oligo dT 18,1 28,8 16,6 15,9 28,4 GSP 18,8 28,7 16,4 17,4 31,8 mix 19,1 27,9 16,3 29,3 max DCt 1,4 2,3 0,8 1,5 4,4 RT priming method

60
Specificity of specific priming
PCR primers used b-tubulin CaVID GAPDH Insulin II Glut 2 18,8 28,7 19 30,6 27 18,7 19,9 22,8 - 23,4 30,1 16,4 20,1 29,7 23,5 31,6 20 17,4 31 25,8 31,9 22,7 31,8 no RT primer 27,6 33,7 23,6 23,1 32,6 RT primers used NTC ~ 35

61
60ºC 37ºC Algorithm: mfold GAPDH 3’ 24 unpaired bases

62
Comparison of reverse transcriptases
Temp MMLV RNase H- Minus (Promega, Germany) 37 M-MLV (Promega) Avian Myeloblastosis Virus (AMV) (Promega) 37 Improm-II (Promega) Omniscript (Qiagen, Germany) 37 cloned AMV (cAMV) (Invitrogen, Germany) 45 ThermoScript RNase H- (Invitrogen) 50 SuperScript III RNase H- (Invitrogen) 50 Ref: Ståhlberg et al. Comparison of reverse transcriptases in gene expression analysis. Clin.Chem. 50(9); (2004)
 37. M-MLV (Promega) 45. Avian Myeloblastosis Virus (AMV) (Promega) 37. Improm-II (Promega) 45. Omniscript (Qiagen, Germany) 37. cloned AMV (cAMV) (Invitrogen, Germany) 45. ThermoScript RNase H- (Invitrogen) 50. SuperScript III RNase H- (Invitrogen) 50. Ref: Ståhlberg et al. Comparison of reverse transcriptases in gene expression analysis. Clin.Chem. 50(9); (2004)")
63
100 – fold variation in RT yield

64
8 transcriptases tested on 6 genes

65
Experimental design to study linearity

66
Effect of carrier - tRNA + tRNA

67
Effect of carrier - tRNA + tRNA

68
RNA dilutions Oligo(dT) Random Hexamers Water Yeast tRNA
Fundera på att ha på flera sen sammanfattande. 68

69
Conclusions The RT reaction shows higher technical variability than QPCR There is no optimum priming strategy Gene specific primers must target accessible regions The RT yield changes over 100-fold with the choice of reverse transcriptase The yield variation is gene specific RT yield is proportional to the amount of template in presence of proper carrier Typical RT yield is % RT-QPCR is highly reproducible as long as the same protocol and reaction conditions are used The efficiency of the RT reaction varies from gene to gene and depends on the conditions – run the RT of all samples using exactly the same protocol and reagents under the same conditions

Similar presentations






>")
 primer Sequence specific primer 1 st strand cDNA.>")





 MCB7300.>")



 Types of Methods: differential solubility.>")



 DNA isolation & purification: –Sample: nucleated cells –Principle: A- PURIFICATION STEPS: 1.Cell lysis 2.Removal of.>")


