Download presentation
Presentation is loading. Please wait.

1
Congenital defects of carbohydrate metobolism

3
CLASSICAL GALACTOSEMIA
Defective enzyme: Galactose-1-phosphate uridyltransferase (incidence 1:40,000) Mechanism: Accumulation of galactose-1-phosphate causes intracellular ATP depletion and very toxic for liver, kidney and brain.
 Mechanism: Accumulation of galactose-1-phosphate causes intracellular ATP depletion and very toxic for liver, kidney and brain.")
4
CLASSICAL GALACTOSEMIA
Galactitol galaktokinase Galactose-1P Galactose-1-phosphate uridyltransferase epimerase Glucose

5
Clinical findings: Progressive symptoms after start of milk feeds
Clinical findings: Progressive symptoms after start of milk feeds. Usually starting on the 3rd and 4th day. Vomiting, diarrhea, jaundice, disturbances of liver function tests, death from severe liver and renal failure, sepsis (especially by E. Coli), bilateral cataracts (due to galactitol accumulation)
, bilateral cataracts (due to galactitol accumulation)")
6
Diagnosis Low galactose-1-phosphate uridyltransferase activity (Beutler test) High galactose and galactose-1-phosphate levels (N: mg/dL) in serum or erythrocytes. Renal tubular damage: Generalized aminoaciduria, albuminuria, Reducing substances in urine (Fehling or Benedict test) due to glucosuria and galactosuria.
 in serum or erythrocytes. Renal tubular damage: Generalized aminoaciduria, albuminuria, Reducing substances in urine (Fehling or Benedict test) due to glucosuria and galactosuria.")
7
Therapy: Lactose-free and galactose restricted diet throughout life
Therapy: Lactose-free and galactose restricted diet throughout life. Keep galactose-1-phosphate levels under 5mg/dL. Complications: Mild mental retardation, ataxia, tremor; gonodal dysfunction, disturbed pubertal development (especially in girls)
")
8
EPIMERASE DEFICIENCY Galactose Galactose-1P Glucose Galactitol
galaktokinase Galactose-1P Galactose-1-phosphate uridyltransferase epimerase Glucose Most of the cases are asymptomatic. Symptomatic cases like classical galactosemia.

9
GALACTOKINASE DEFICIENCY
Galactose Galactitol galaktokinase Galactose-1P Galactose-1-phosphate uridyltransferase epimerase Glucose

10
GALACTOKINASE DEFICIENCY
Clinical findings Rapidly progressing bilateral central cataract. Reversible in the first few weeks of life. No hepatorenal damage Therapy: Lactose-free diet

11
HEREDITARY FRUCTOSE INTOLERANCE
Fructokinase Fructose-1P Aldolase Glucose

12
HEREDITARY FRUCTOSE INTOLERANCE
Defective enzyme: Aldolase B (incidence 1:20,000) Mechanism: Accumulation of fructose-1-phosphate causes intracellular ATP depletion and very toxic for liver, kidney and brain.
 Mechanism: Accumulation of fructose-1-phosphate causes intracellular ATP depletion and very toxic for liver, kidney and brain.")
13
Clinical findings: Symptoms after weaning or supplementary feeds.
Vomiting, apathy liver dysfunction with hepatomegaly, hypoglycemia (inhibition of glycogenolysis) renal tubular dysfunction, distribution of liver function tests, failure to thrive, aversion to fructose containing foods/sweets, no caries.
 renal tubular dysfunction, distribution of liver function tests, failure to thrive, aversion to fructose containing foods/sweets, no caries.")
14
Diagnosis: Clinical symptoms with fructose loading (dangerous
Diagnosis: Clinical symptoms with fructose loading (dangerous!); positive effect of withdrawing fructose; enzyme studies. Renal tubular damage: Generalized aminoaciduria, albuminuria, Reducing substances in urine (Fehling or Benedict test) due to glucosuria and galactosuria. Therapy: Strict fructose restricted diet throughout life.
; positive effect of withdrawing fructose; enzyme studies. Renal tubular damage: Generalized aminoaciduria, albuminuria, Reducing substances in urine (Fehling or Benedict test) due to glucosuria and galactosuria. Therapy: Strict fructose restricted diet throughout life.")
15
ESSENTIAL FRUCTOSURIA
Defective enzyme: Fructokinase Clinical findings: asymptomatic. Diagnosis: Positive reducing substance in urine (incidental finding)
")
16
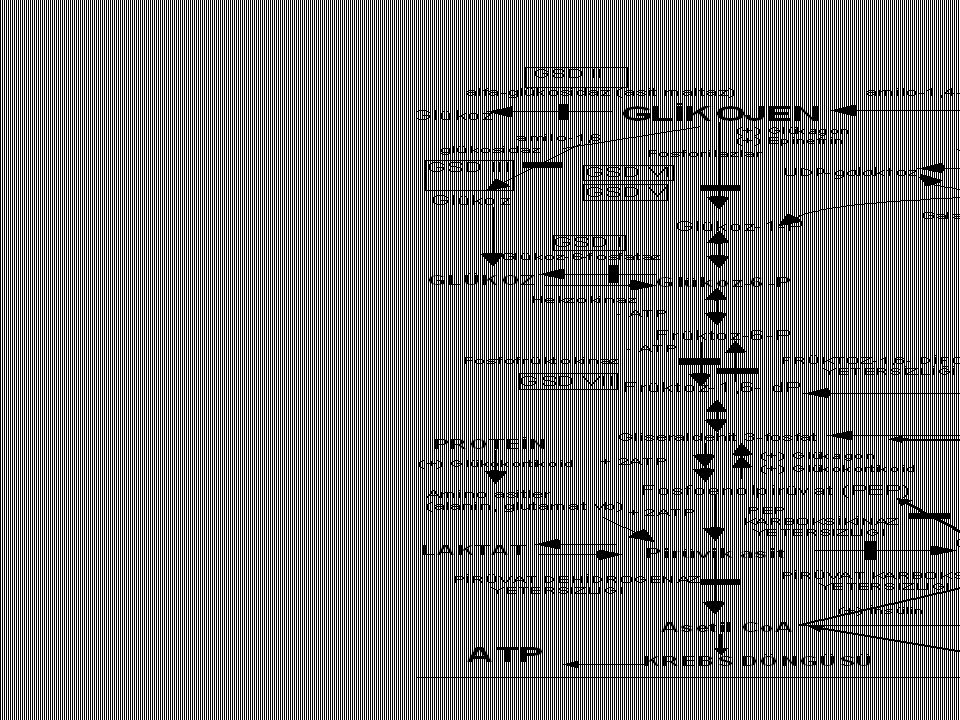
Glycogen storage diseases

18
Glycogen storage diseases – Classification
Liver involvement and hypoglycemia (Type 0, I, VI) Muscle involvement (Type II, V, VII) Liver involvement, muscle involvement and hypoglycemia (Type III). Liver cirrhosis: (Type III , Type IV).
 Muscle involvement (Type II, V, VII) Liver involvement, muscle involvement and hypoglycemia (Type III). Liver cirrhosis: (Type III , Type IV).")
19
GSD Type O Defect: Liver glycogen synthase
Clinical: Convulsions in the early hours of morning No Hepatomegaly Hypoglycemia inresponsive to glucagon

21
Type I a (von Gierke)/ Clinic/Laboratory
Neonatal hypoglycemia Lactic acidosis, ketosis Hepatomegaly Hyperuricemia gout Hyperlipidemia Lipid accumulation in the cheeks (doll facies).
.")
22
Type I a (von Gierke)/Diagnosis
Hypoglycemia inresponsive to glucagon Glucose challange test: Glucose (increase), Lactate (decrease) Diagnosis: Low liver glucose-6-phosphatase levels.
, Lactate (decrease) Diagnosis: Low liver glucose-6-phosphatase levels.")
23
GSD Type I a (von Gierke)/Therapy
Avoid hypoglycemia by means of continous carbohydrate intake. Frequent meals (every 2-3 hours in infants, 4-6 hours in school age) Slowly resorbed carbohydrates (raw –uncooked- starch) Limited fructose, lactose, no sucrose, Nights: Continous intake of glucose polymers (starch) via nasogastric tube
 Slowly resorbed carbohydrates (raw –uncooked- starch) Limited fructose, lactose, no sucrose, Nights: Continous intake of glucose polymers (starch) via nasogastric tube.")
24
GSD Type Ib Enzyme: Glucose-6-phosphate transporter (edoplasmic reticulum) Clinical: looks like GSD type Ia Plus: neutropenia, leucocyte dysfunction, frequent infections, Crohn like inflammatory bowel disease. Treatment of neutropenia: G-CSF and GM-CSF

25
GSD type Ic and Id Clinical: Similar GSD type Ia
Ic: Inorganic phospate transport defect (T2 protein disorder) Id: GLUT 7 microsomal transport defect
 Id: GLUT 7 microsomal transport defect.")
26
GSD type VI Hers disease
Enzyme: Liver phosphorylase defect Clinical: Failure to thrive, hepatomegaly, mild hypoglycemia Diagnosis: Elevated lactate and transaminases, enzyme studies: Liver, erythrocytes, leucocytes Prognosis: Fairly good, often asymptomatic

28
GSD type VII Tarui disease
Enzyme: phosphofruktokinase (muscle, erythrocyte, platelets) Severe myopathy Postprandial exercize intolerance (does not respond to glucose infusion) Mild hemolytic anemia Hyperuricemia (post exercize) PAS positive, abnormal amilopectin accumulation
 Severe myopathy. Postprandial exercize intolerance (does not respond to glucose infusion) Mild hemolytic anemia. Hyperuricemia (post exercize) PAS positive, abnormal amilopectin accumulation.")
29
GSD type IX Enzyme: Phosphorylase kinase
X related liver form (most frequent) AR liver and muscle form AR liver form Muscle form Cardiac form
 AR liver and muscle form. AR liver form. Muscle form. Cardiac form.")
30
GSD type II (Pompe disease)
Enzyme: alpha-1,4-glucosidase (asid maltase); lysosomal enzyme; minimal role in glucose metabolism. Storage of cholesterol esters and triglycerides. İnfantile form : Severe cardiomyopathy and myopathy, hypotonia, hepatomegaly, failure to thrive, untreated fatal in the first year Juvenile-adult form: Slowly progressing skeletal muscle weakness, no cardiomyopathy Diagnosis; Vacuolated lymphocytes, EKG: PR-shortness and massive QRS waves, enzyme studies Enzyme replacement therapy
; lysosomal enzyme; minimal role in glucose metabolism. Storage of cholesterol esters and triglycerides. İnfantile form : Severe cardiomyopathy and myopathy, hypotonia, hepatomegaly, failure to thrive, untreated fatal in the first year. Juvenile-adult form: Slowly progressing skeletal muscle weakness, no cardiomyopathy. Diagnosis; Vacuolated lymphocytes, EKG: PR-shortness and massive QRS waves, enzyme studies. Enzyme replacement therapy.")
32
GSDType III (Cori / Forbes disease)
Defective Enzyme: Debrancher enzyme= amylo-1, 6-glucosidase accumulation of limit dextrin in liver and muscle. Type IIIa: (Liver+muscle involvement): In infancy same as in GSD Type I; but normal lactate/uric acid, mild hypoglycemia, improvement in childhood, hepatomegaly, normal kidney size, failure to thrive; Progressive myopathy, cardiomyopathy, neuropathy Type IIIb: (Only liver involvement) Diagnosis: decreased alanine, leucine, isoleucine, elevated valine; increased transaminases and cholesterol. Enzyme studies: Liver, muscle, erythrocytes, fibroblasts Therapy: Frequent feeding, maintain normoglycemia
: In infancy same as in GSD Type I; but normal lactate/uric acid, mild hypoglycemia, improvement in childhood, hepatomegaly, normal kidney size, failure to thrive; Progressive myopathy, cardiomyopathy, neuropathy. Type IIIb: (Only liver involvement) Diagnosis: decreased alanine, leucine, isoleucine, elevated valine; increased transaminases and cholesterol. Enzyme studies: Liver, muscle, erythrocytes, fibroblasts. Therapy: Frequent feeding, maintain normoglycemia.")
33
GSD Type IV (Andersen disease)
Defective Enzyme: Branching enzyme = amylo-1,4-> 1,6 transglucosidase; accumulation of amylopectin like glycogen molecule Classic form: failure to thrive, hepatomegaly, progressive liver disease -> cirhossis, liver failure, often fatal by age of 4-5 years Mild form; Non-progressive Neuromuscular forms Enzyme studies: Liver, muscle, erythrocytes, fibroblasts Therapy: Liver Transplantation ?

35
GSD Type V (McArdle disease)
Enzyme: Muscle phosphorylase Clinical: Adolescent/Adult onset; exercise intolerance, fatigue, muscle cramps, myoglobinuria No postischemic hyperlactatemia, elevated CK Diagnosis: muscle biopsy Therapy: Avoid exessive exercise, creatine supplementation

36
Glucose transport defects

37
GLUT 1 deficiency Clinical: Epileptic encephalopathy, microcephaly, psyhomotor retardation Diagnosis: Fasting CSF: CSF glucose/ Blood glucose<0.45; lactate: N, or decreased Therapy: Ketogenic diet. Avoid these drugs: barbiturates, ethanol, methylxantines Prognosis: Satisfactory with early treatment

38
GSD tip XI (Fanconi-Bickel syndrome)
GLUT 2 defect Clinically resembles GSD type Ia Dwarfism, Fanconi syndrome, rickets Fasting hypoglycemia (no response to glucagon), postprandial hyperglycemia, aminoaciduria, phosphaturia, glucosuria Glycogen accumulation in liver and kidneys
, postprandial hyperglycemia, aminoaciduria, phosphaturia, glucosuria. Glycogen accumulation in liver and kidneys.")
39
Congenital glucose-galactose malabsorption
SGLT1 deficiency Severe intestinal glucose and galactose transport defects, mild renal transport defect. Clinical: Severe neonatal diarrhoea, dehydration Diagnosis: Mild glucosuria, reducing substance in stools, normal fructose tolerance Therapy: Dietary replacement of gucose/galactose with fructose

40
Renal glucosuria SGLT2 deficiency
Renal transport defect of glucose and galactose No intestinal defect Benign glucosuria with normal blood glucose and absence of generalized tubular dysfunction. Therapy: None

41
CYSTIC FIBROSIS

42
Kistik fibrozda solunum epitelinde görülen sıvı sekresyonu ve emilimindeki bozukluklar

43
Ter sekresyonu

44
Cystic fibrosis-Genetics
Cystic fibrosis gene is localized in the long arm of 7th chromosome (7q31.2). Cystic fibrosis gene codes cystic fibrosis transmembrane regulator (CFTR). There are more than 600 mutations. The most frequent mutation is delta-F508, 70-80% in USA and 30% in Turkey) Incidence: 1:2500 in Caucasians, less frequent in Orientals where cholera is endemic. Incidence in Turkey is not known, but presumed to be high.
. Cystic fibrosis gene codes cystic fibrosis transmembrane regulator (CFTR). There are more than 600 mutations. The most frequent mutation is delta-F508, 70-80% in USA and 30% in Turkey) Incidence: 1:2500 in Caucasians, less frequent in Orientals where cholera is endemic. Incidence in Turkey is not known, but presumed to be high.")
45
PATHOGENESIS CTFR is inserted in the cell membranes and has the properties of ion channels. In cystic fibrosis cAMP-dependent chloride channels that regulate the water secretion and sodium channels regulate the water reabsorption are defective.

46
Apical membranes of epithelial cells of exocrine glands are impermeable to chloride, and secondarily sodium and water. Additionally epithelial cells reabsorb excessive amount amounts of sodium and secondarily water. Decreased water secretion and increased water reabsorption (dehydrated secretions) lead obstruction, cysts and fibrosis in the exocrine glands (bronchial tree, pancreatic duct, gastrointestinal glands etc) with the exception of sweat gland.
 lead obstruction, cysts and fibrosis in the exocrine glands (bronchial tree, pancreatic duct, gastrointestinal glands etc) with the exception of sweat gland.")
47
In sweat glands, chloride secretion and thereby the water secretion is normal in the secretory coil.
On the other hand sodium and secondarily chloride reabsorption by the ductus is decreased. Therefore excessive amounts of salt are lost in the sweat (chloride levels are more than 60 mEq/L).
.")
48
Kistik fibroz: Patogenez

49
CLINICAL MANIFESTATIONS
The most life threatening clinical features of CF, are pulmonary obstruction and infections. Thick mucous secretions are associated with chronic obstructive lung disease predominantly involving the small airways. Recurrent and persistent infections with Pseudomonas aeroginosa and cepecia (virtually diagnostic) and Staphylococcous lead to bronchiectasis and respiratory failure, often accompanied by cor pulmonale and death
 and Staphylococcous lead to bronchiectasis and respiratory failure, often accompanied by cor pulmonale and death.")
50
CLINICAL MANIFESTATIONS
In 80-90% of the patients exocrine pancreatic deficiency causes steatorhea and failure to thrive. In 10-20% of the patients, meconium ileus occurs in the neonatal period. Failure to pass meconium in the first hours of life. Abdominal roentgenograms show dilated loops of bowel with a collection of granular ground-glass material in the lower central abdomen.

51
Meconium ileus

52
CLINICAL MANIFESTATIONS
Excessive lost of salt in the sweat leads hyponatremic dehydration in young children especially during high environmental temperature. Secondary hyperaldosteronism causes hypopotasemia and metabolic alkalosis (Pseudo-Bartter syndrome). 10% of patients diabetes mellitus develops, especially after the age of ten.
. 10% of patients diabetes mellitus develops, especially after the age of ten.")
53
CRITERIA FOR DIAGNOSIS OF CYSTIC FIBROSIS
Typical pulmonary manifestations and/or Typical gastrointestinal manifestations and/or A history of cystic fibrosis in the family plus * Sweat chloride concentration >60mEq/L and/or * Pathologic CTFR mutation on both chromosomes.

54
NEONATAL SCREENING Most of the patients with CF can be identified by determination of immunoreactive tripsinogen in blood spots (Guthrie card). This test is neither sufficiently sensitive nor spesific for CF. This test is not valid after the age of three months (Serum tripsinogen levels decrease during progressive pancreatic damage).
.")
55
Pulmonary therapy The objective of pulmonary therapy is to clear secretions from airways and to control infections Chest percussion + postural drainage Bronchodilators: ß-sympathomimetic agents, teophylline Expectorants: Are not effective, curcumin may be helpful (stimulation of Cl channel) %3-7 NaCl inhalation
 %3-7 NaCl inhalation.")
56
Antibiotic therapy Aerosol and/or intravenous: (Pseudomonas is more difficult to treat and rarely is eradicated) Pseudomonas aeroginosa: amikasin, gentamycin, tobramycin, netilmycin, imipenem Staphylococcous aereus: voncomycin, oxacillin

57
Nutritional therapy High caloric diet (150% of normal calorie) without fat restriction Pancreatic enzyme replacement Fat-soluble vitamins (A,D,E,K) supplementation
 supplementation.")
Similar presentations













![Islets of Langerhan. Prof. K. Sivapalan. 08-01-14Islets of Langerhan2 Histology. A cells 20 % [glucogon] B cells 50% [Insulin] D cells 8% [somatostatin]](/15/4663650/big_thumb.jpg "Islets of Langerhan. Prof. K. Sivapalan. 08-01-14Islets of Langerhan2 Histology. A cells 20 % [glucogon] B cells 50% [Insulin] D cells 8% [somatostatin]>")










