Download presentation
Presentation is loading. Please wait.

1
Pharmacotherapy of Cardiovascular Diseases
Myocardial Infarction, Hypertension, Congestive Heart Failure , Heperlipidemia.

2
Novel mechanism for cell fate regulation in human vascular and blood cells
Fig: Cardiovascular disease and cell fate of the human vascular and blood cells.

3
Figure 2: Cell fate control in the human vascular and blood cells.

4
Elucidations of the regulatory and pathophysiological molecular mechanisms of control systems for cell fate in the human cells yield information regarding the drug action, metabolism and the target of therapeutic drug discovery. The cell surface GPCR (G-protein-coupled receptor (IP)) and nuclear receptor PPAR (peroxisome proliferator-activated receptor (delta)), a regulator of energy metabolism and a key target for obesity and adipogenesis, have important roles in regulation of cell fate in a cooperative and/or competitive manner. The new methods for regulation of human vascular and blood cells via related signaling pathways.
) and nuclear receptor PPAR (peroxisome proliferator-activated receptor (delta)), a regulator of energy metabolism and a key target for obesity and adipogenesis, have important roles in regulation of cell fate in a cooperative and/or competitive manner. The new methods for regulation of human vascular and blood cells via related signaling pathways..")
5
Myocardial Infarction
Drugs used in Anginal Pain: (1) Organic Nitrates – *Glyceryl Trinitrates (GTN) / Nitroglycerin. (2) Calcium Channel Blockers- * Verapamil, * Nifedipine. (3) Beta-adrenoceptor Blockers- * Atenolol, * Propanolol. (4) Dipyridamol.
 Organic Nitrates – *Glyceryl Trinitrates (GTN) / Nitroglycerin. (2) Calcium Channel Blockers- * Verapamil, * Nifedipine. (3) Beta-adrenoceptor Blockers- * Atenolol, * Propanolol. (4) Dipyridamol.")
6
M/A of Organic Nitrates:
Organic Nitrates act solely by relaxing smooth muscle of blood vessels by the following way. (1) Denitration of organic nitrates. (Organic Nitrates- Inorganic Nitrates). (2) Inorganic nitrates are converted to NO (Such as EDRF). (3) Activation of Guanylyl Cyclase (GC) by NO. (4) Increase formation of cGMP. (5) cGMP reduces intracellular Ca+++ conc. (6) Relaxation of vasular smooth muscle, Increase coronary flow. Generalized vasodilation: Decrease Blood Pressure. (7) Improve Myocardial perfusion. Reduce myocardial oxygen demand. EDRF means Endothelial derived relaxant factor. It causes vasodilation.
 Denitration of organic nitrates. (Organic Nitrates- Inorganic Nitrates). (2) Inorganic nitrates are converted to NO (Such as EDRF). (3) Activation of Guanylyl Cyclase (GC) by NO. (4) Increase formation of cGMP. (5) cGMP reduces intracellular Ca+++ conc. (6) Relaxation of vasular smooth muscle, Increase coronary flow. Generalized vasodilation: Decrease Blood Pressure. (7) Improve Myocardial perfusion. Reduce myocardial oxygen demand. EDRF means Endothelial derived relaxant factor. It causes vasodilation.")
7
History of Nitroglycerin
Nitroglycerin, which was originally synthesized by Ascanio Sobrero, was used by Alfred Nobel to manufacture dynamite. It was in Nobel's dynamite factories in the late 1860s that the antianginal effect of nitroglycerin was discovered. Two interesting observations were made. First, factory workers on Monday mornings often complained of headaches that disappeared over the weekends. Second, factory workers suffering from angina pectoris or heart failure often experienced relief from chest pain during the work week, but which recurred on weekends. Both effects were attributed to the vasodilator action of nitroglycerin, which quickly became apparent to the physicians and physiologists in local communities. In the late 1970s and early 1980s, the vasodilator effect of nitroglycerin was discovered to be caused by nitric oxide (NO), which was apparently generated from nitroglycerin in vascular smooth muscle, “Nitric Oxide as a Signaling Molecule in the Cardiovascular System”.
, which was apparently generated from nitroglycerin in vascular smooth muscle, Nitric Oxide as a Signaling Molecule in the Cardiovascular System .")
8
Molecular Mechanism of nitroglycerin
Nitroglycerin (GTN), often used in conditions of cardiovascular ischaemia, acts through the liberation of nitric oxide (NO) and the local concentration of NO in the tissue is responsible for any biological effect. However, little is known about the way in which the concentration of NO from GTN and other NO-donors is influenced by low oxygen tension in the target tissues. The biological effect of NO is dependent of the concentration at the site of action. under hypoxic conditions the endogenous, L-arginine dependent NO synthesis may be impaired. Even the generation of ‘exogenous' NO from different NO-donors is likely to be sensitive to alterations in tissue PO2. Nitrite (NO2−) is a metabolite of organic nitrates. Therefore, the increase of NO from GTN during hypoxia might be due to increased indirect metabolism of GTN through the conversion of NO2− to NO. To evaluate this possibility we infused increasing concentrations of inorganic NO2− and NO3− in buffer-perfused lungs during hypoxic conditions. The mechanism for increase of GTN-derived NO during hypoxia was via increased conversion of NO2− to NO. The mitochondrial aldehyde dehydrogenase (ALDH2, mtALDH) was recently found to catalyze the reduction of nitroglycerin (glyceryl trinitrate [GTN]) to generate nitrite and 1,2-glyceryl dinitrate. The nitrite generated within the mitochondria is metabolized further to generate nitric oxide (NO)-based bioactivity, by reduction to NO and/or by conversion to S-nitrosothiol, as revealed by a series of biochemical, pharmacologic, and genetic studies.
, often used in conditions of cardiovascular ischaemia, acts through the liberation of nitric oxide (NO) and the local concentration of NO in the tissue is responsible for any biological effect. However, little is known about the way in which the concentration of NO from GTN and other NO-donors is influenced by low oxygen tension in the target tissues. The biological effect of NO is dependent of the concentration at the site of action. under hypoxic conditions the endogenous, L-arginine dependent NO synthesis may be impaired. Even the generation of ‘exogenous NO from different NO-donors is likely to be sensitive to alterations in tissue PO2. Nitrite (NO2−) is a metabolite of organic nitrates. Therefore, the increase of NO from GTN during hypoxia might be due to increased indirect metabolism of GTN through the conversion of NO2− to NO. To evaluate this possibility we infused increasing concentrations of inorganic NO2− and NO3− in buffer-perfused lungs during hypoxic conditions. The mechanism for increase of GTN-derived NO during hypoxia was via increased conversion of NO2− to NO. The mitochondrial aldehyde dehydrogenase (ALDH2, mtALDH) was recently found to catalyze the reduction of nitroglycerin (glyceryl trinitrate [GTN]) to generate nitrite and 1,2-glyceryl dinitrate. The nitrite generated within the mitochondria is metabolized further to generate nitric oxide (NO)-based bioactivity, by reduction to NO and/or by conversion to S-nitrosothiol, as revealed by a series of biochemical, pharmacologic, and genetic studies.")
9
Nitric oxide (NO), a molecule produced by many cells in the body, and has several important actions. In the cardiovascular system, NO is primarily produced by vascular endothelial cells. This endothelial-derived NO has several important functions including relaxing vascular smooth muscle (vasodilation), inhibiting platelet aggregation (anti-thrombotic), and inhibiting leukocyte-endothelial interactions (anti-inflammatory). These actions involve NO-stimulated formation of cGMP. Nitrodilators are drugs that mimic the actions of endogenous NO by releasing NO or forming NO within tissues. These drugs act directly on the vascular smooth muscle to cause relaxation and therefore serve as endothelial-independent vasodilators.
, inhibiting platelet aggregation (anti-thrombotic), and inhibiting leukocyte-endothelial interactions (anti-inflammatory). These actions involve NO-stimulated formation of cGMP. Nitrodilators are drugs that mimic the actions of endogenous NO by releasing NO or forming NO within tissues. These drugs act directly on the vascular smooth muscle to cause relaxation and therefore serve as endothelial-independent vasodilators..")
10
There are two basic types of nitrodilators: those that release NO spontaneously (e.g., sodium nitroprusside) and organic nitrates that require an enzymatic process to form NO. Organic nitrates do not directly release NO, however, their nitrate groups interact with enzymes and intracellular sulfhydryl groups that reduce the nitrate groups to NO or to S-nitrosothiol, which then is reduced to NO. Nitric oxide activates smooth muscle soluble guanylyl cyclase (GC) to form cGMP. Increased intracellular cGMP inhibits calcium entry into the cell, thereby decreasing intracellular calcium concentrations and causing smooth muscle relaxation. NO also activates K+ channels, which leads to hyperpolarization and relaxation. Finally, NO acting through cGMP can stimulate a cGMP-dependent protein kinase that activates myosin light chain phosphatase, the enzyme that dephosphorylates myosin light chains, which leads to relaxation. 1. Vasodilation by releasing NO in blood vessels which causes relaxation of smooth muscle -> Decrease venus return -> Decrease preload -> Decrease oxygen demand -> Pain is relieved. 2. Also causes arterial dilatation -> Decrease arterial resistance -> Decrease afterload -> Decrease oxygen demand -> Pain is relieved.
 to form cGMP. Increased intracellular cGMP inhibits calcium entry into the cell, thereby decreasing intracellular calcium concentrations and causing smooth muscle relaxation. NO also activates K+ channels, which leads to hyperpolarization and relaxation. Finally, NO acting through cGMP can stimulate a cGMP-dependent protein kinase that activates myosin light chain phosphatase, the enzyme that dephosphorylates myosin light chains, which leads to relaxation. 1. Vasodilation by releasing NO in blood vessels which causes relaxation of smooth muscle -> Decrease venus return -> Decrease preload -> Decrease oxygen demand -> Pain is relieved. 2. Also causes arterial dilatation -> Decrease arterial resistance -> Decrease afterload -> Decrease oxygen demand -> Pain is relieved.")
11
Organic nitrates can dilate both arteries and veins, venous dilation predominates when these drugs are given at normal therapeutic doses. Venous dilation reduces venous pressure and decreases ventricular preload. This reduces ventricular wall stress and oxygen demand by the heart, thereby enhancing the oxygen supply/demand ratio. A reduction in preload (reduced diastolic wall stress) also helps to improve subendocardial blood flow, which is often compromised in coronary artery disease. Mild coronary dilation or reversal of coronary vasospasm will further enhance the oxygen supply/demand ratio and diminish the anginal pain. Coronary dilation occurs primarily in the large epicardial vessels, which diminishes the likelihood of coronary vascular steal. Systemic arterial dilation reduces afterload, which can enhance cardiac output while at the same time reducing ventricular wall stress and oxygen demand. At high concentrations, excessive systemic vasodilation may lead to hypotension and a baroreceptor reflex that produces tachycardia. When this occurs, the beneficial effects on the oxygen supply/demand ratio are partially offset. Furthermore, tachycardia, by reducing the duration of diastole, decreases the time available for coronary perfusion, most of which occurs during diastole.
 also helps to improve subendocardial blood flow, which is often compromised in coronary artery disease. Mild coronary dilation or reversal of coronary vasospasm will further enhance the oxygen supply/demand ratio and diminish the anginal pain. Coronary dilation occurs primarily in the large epicardial vessels, which diminishes the likelihood of coronary vascular steal. Systemic arterial dilation reduces afterload, which can enhance cardiac output while at the same time reducing ventricular wall stress and oxygen demand. At high concentrations, excessive systemic vasodilation may lead to hypotension and a baroreceptor reflex that produces tachycardia. When this occurs, the beneficial effects on the oxygen supply/demand ratio are partially offset. Furthermore, tachycardia, by reducing the duration of diastole, decreases the time available for coronary perfusion, most of which occurs during diastole.")
12
Calcium Channel Blockers
Ca++ is required for: (1) Cardiac contraction, (2) Smooth muscle contraction, (3) Propagation of cardiac impulse. Main Calcium Antagonists are- Nifedipine ( relative smooth muscle selective), Amlodipine (“), Verapamil (relatively cardioselective), Diltiazem (intermediate in action).
 Cardiac contraction, (2) Smooth muscle contraction, (3) Propagation of cardiac impulse. Main Calcium Antagonists are- Nifedipine ( relative smooth muscle selective), Amlodipine ( ), Verapamil (relatively cardioselective), Diltiazem (intermediate in action).")
13
M/A of Calcium Channel Blockers
Ca++ channel blockers -> Binds with voltage dependent Ca++ channel in depolarized membrane. (The drugs act from inner side of the membrane). Decrease in transmembrane Ca++ current -> Smooth muscle: relaxation Heart: negative ionotropic action.
. Decrease in transmembrane Ca++ current -> Smooth muscle: relaxation. Heart: negative ionotropic action.")
14
Flow chart: Ca++ channel Blockers (-) Ca++ Calmodulin
Ca++ calmodulin complex. Active MLCK Myosin- LC Kinase (MLCK) Myosin light chain (MLC) Myosin LC-PO MLC Actin Contraction Relaxation
 Myosin light chain (MLC) Myosin LC-PO MLC. Actin. Contraction Relaxation.")
15
Molecular mechanism of Calcium Channel blockers
L-type channels control voltage-dependent Ca2+ influx into cardiac and vascular smooth muscle, channel blockers inhibit depolarization-induced Ca2+ entry into muscle cells in the cardiovascular system. This causes a decrease in blood pressure, reduced cardiac contractility (and hence oxygen consumption) and antiarrhythmic effects. Therefore these drugs are used clinically to treat hypertension, myocardial ischemia and cardiac arrhythmias. Ca2+ channel block by calcium antagonists: Like other voltage-gated cation channels, Ca2+ channels exist in at least three states. A resting state stabilized at negative potentials (such as the resting potentials of most electrically excitable cells) which is a closed state from which the channel can open. The open state is induced by depolarization. Channels do not stay open indefinitely because they are “turned off” during prologend depolarization by transition into an inactivated state. Once the cell repolarizes inactivated channels return to the resting state and are now again available for opening. Ca2+ channel blockers inhibit Ca2+ flux mainly by “allosterically” stabilizing the inactivated closed state. By delaying its transition to the resting state after repolarization some blockers can also increase the refractory period of these channels.
 and antiarrhythmic effects. Therefore these drugs are used clinically to treat hypertension, myocardial ischemia and cardiac arrhythmias. Ca2+ channel block by calcium antagonists: Like other voltage-gated cation channels, Ca2+ channels exist in at least three states. A resting state stabilized at negative potentials (such as the resting potentials of most electrically excitable cells) which is a closed state from which the channel can open. The open state is induced by depolarization. Channels do not stay open indefinitely because they are turned off during prologend depolarization by transition into an inactivated state. Once the cell repolarizes inactivated channels return to the resting state and are now again available for opening. Ca2+ channel blockers inhibit Ca2+ flux mainly by allosterically stabilizing the inactivated closed state. By delaying its transition to the resting state after repolarization some blockers can also increase the refractory period of these channels.")
17
From the fig. Simplified view of the pharmacological action of L-type Ca2+ channel blockers in arterial smooth muscle: In contrast to cardiomyocytes action potentials are not carried by fast sodium channels in smooth muscle and depolarzations are more long lasting. Contraction requires the binding of Ca2+ to calmodulin, which then activates myosin light chain kinase (MLCK). MLCK phosphorylates the light chain of myosin which turns on contraction. The Ca2+ for activation of this pathway can enter through L-type Ca2+ channels in response to depolarization. Ca2+ channel blockers inhibit this pathway through concentration-dependent block of Ca2+ entry. Alternatively, Ca2+ can be released from intracellular stores after activation of membrane receptors (e.g. of angiotensin II AT1 or a1 -adrenergic receptors) coupled to IP3 production. IP3 opens IP3 receptor channels, RyR related Ca2+ release channels in the SR. This process does not involve L-type Ca2+ channels and is not inhibited by Ca2+ channel blockers. Store-depletion also triggers the activation of "store-operated channels" (SOC) in the plasma membrane which are also not sensitive to Ca2+ channel blockers. Receptor-mediated activation of cAMP-dependent protein kinase (cAMP-PK) results in muscle relaxation through different mechanisms. D1-R, dopamine1 receptor; AR, adrenergic receptor; PLC, phospholipase C. In another way, by blocking L-type channels in arterial smooth muscle they reduce Ca2+ influx during depolarisation. Thus less Ca2+ is available for activation of calmodulin which activates myosin-light chain kinase and thereby turns on actin-myosin interaction. Note that smooth muscle also contracts after stimulation of receptor-activated pathways. Agonists of angiotensin AT1 (e.g. angiotensin II) and a1 -adrenergic receptors (e.g. noradrenaline) release Ca2+ from intracellular IP3-sensitive stores. Noradrenaline-induced contractions are much less sensitive to Ca2+ channel blockers. The differential contribution of depolarization-induced and receptor-activated contraction in different types of smooth muscle and under different pathophysiological conditions is one of the explanations why Ca2+ channel blockers are not effective relaxants in other diseases (such as e.g. of bronchial muscle in asthma or uretral spasms).
 and a1 -adrenergic receptors (e.g. noradrenaline) release Ca2+ from intracellular IP3-sensitive stores. Noradrenaline-induced contractions are much less sensitive to Ca2+ channel blockers. The differential contribution of depolarization-induced and receptor-activated contraction in different types of smooth muscle and under different pathophysiological conditions is one of the explanations why Ca2+ channel blockers are not effective relaxants in other diseases (such as e.g. of bronchial muscle in asthma or uretral spasms).")
19
Simplified view of the pharmacological action of L-type Ca2+ channel blockers in cardiac myocytes: In cardiac myocytes L-type Ca2+ channels open when the plasma membrane is depolarised by an action potential carried along the muscle cells by the opening of voltage-gated sodium-channels (Na-Ch). The action potential is terminated (an its duration is determined) by the opening of potassium channels (K-Ch). Ca2+ influx triggers massive release of Ca2+ from intracellular stores by opening ryanodine-sensitive Ca2+ channels (ryanodine receptors, RyR) in the sarcoplasmic reticulum, resulting in an intracellular Ca2+ transient. Ca2+ influx and released Ca2+ directly initiate contraction. Contraction is terminated by the rapid uptake into the SR by SR Ca2+ ATPases (SERCA). b-adrenergic receptor stimulation increases inotropy by phosphorylation (P) of phospholamban (PLN) and L-type channels through cAMP-dependent protein kinase (cAMP-PK). The resulting stimulation of Ca2+ influx and Ca2+ - pump activity increases the load of Ca2+ in the SR stores. This leads to enhanced Ca2+ transients upon depolarization. Inhibition of Ca2+ influx through L-type Ca2+ channels by Ca2+ channel blockers causes decreased Ca2+ entry and SR load. Less Ca2+ influx and release result in smaller Ca2+ transients and a decrease in contractile force. In the heart calcium entering through L-type channels during the action potential serves as a trigger ("trigger calcium") for further calcium release from the sarcoplasmic reticulum which initiates contraction. b-adrenergic receptor activation increases inotropy at least in part by cAMP-dependent phosphorylation of L-type channels thereby increasing calcium entry.
 for further calcium release from the sarcoplasmic reticulum which initiates contraction. b-adrenergic receptor activation increases inotropy at least in part by cAMP-dependent phosphorylation of L-type channels thereby increasing calcium entry.")
20
Beta-Blockers/ Beta adrenoceptors antagonist
Cardiac Effects Decrease contractility, (negative intropy) Decrease relaxation rate, (negative lusitropy) Decrease heart rate, (negative chronotropy) Decrease conduction velocity, (negative dromotropy). Vascular Effects Smooth muscle contraction, (mild vasoconstriction)
 Decrease relaxation rate, (negative lusitropy) Decrease heart rate, (negative chronotropy) Decrease conduction velocity, (negative dromotropy). Vascular Effects. Smooth muscle contraction, (mild vasoconstriction)")
21
Molecular Mechanism of Beta-adrenoceptor antagonist (Beta-Blockers)
Beta-blockers are drugs that bind to beta-adrenoceptors and thereby block the binding of norepinephrine and epinephrine to these receptors. This inhibits normal sympathetic effects that act through these receptors. Therefore, beta-blockers are sympatholytic drugs. Some beta-blockers, when they bind to the beta-adrenoceptor, partially activate the receptor while preventing norepinephrine from binding to the receptor. These partial agonists therefore provide some "background" of sympathetic activity while preventing normal and enhanced sympathetic activity. These particular beta-blockers (partial agonists) are said to possess intrinsic sympathomimetic activity (ISA). Some beta-blockers also possess what is referred to as membrane stabilizing activity (MSA). This effect is similar to the membrane stabilizing activity of sodium-channels blockers that represent Class I antiarrhythmics. The first generation of beta-blockers were non-selective, meaning that they blocked both beta-1 (β1) and beta-2 (β2) adrenoceptors. Second generation beta-blockers are more cardioselective in that they are relatively selective for β1 adrenoceptors. This relative selectivity can be lost at higher drug doses. Finally, the third generation beta-blockers are drugs that also possess vasodilator actions through blockade of vascular alpha-adrenoceptors.
 are said to possess intrinsic sympathomimetic activity (ISA). Some beta-blockers also possess what is referred to as membrane stabilizing activity (MSA). This effect is similar to the membrane stabilizing activity of sodium-channels blockers that represent Class I antiarrhythmics. The first generation of beta-blockers were non-selective, meaning that they blocked both beta-1 (β1) and beta-2 (β2) adrenoceptors. Second generation beta-blockers are more cardioselective in that they are relatively selective for β1 adrenoceptors. This relative selectivity can be lost at higher drug doses. Finally, the third generation beta-blockers are drugs that also possess vasodilator actions through blockade of vascular alpha-adrenoceptors.")
22
Beta-blockers bind to beta-adrenoceptors located in cardiac nodal tissue, the conducting system, and contracting myocytes. The heart has both β1 and β2 adrenoceptors, although the predominant receptor type in number and function is β1. These receptors primarily bind norepinephrine that is released from sympathetic adrenergic nerves. Additionally, they bind norepinephrine and epinephrine that circulate in the blood. Beta-blockers prevent the normal ligand (norepinephrine or epinephrine) from binding to the beta-adrenoceptor by competing for the binding site.
 from binding to the beta-adrenoceptor by competing for the binding site..")
23
Beta-adrenoceptors are coupled to a Gs-proteins, which activate adenylyl cyclase to form cAMP from ATP. Increased cAMP activates a cAMP-dependent protein kinase (PK-A) that phosphorylates L-type calcium channels, which causes increased calcium entry into the cell. Increased calcium entry during action potentials leads to enhanced release of calcium by the sarcoplasmic reticulum in the heart; these actions increase inotropy (contractility). Gs-protein activation also increases heart rate (chronotropy). PK-A also phosphorylates sites on the sarcoplasmic reticulum, which lead to enhanced release of calcium through the ryanodine receptors (ryanodine-sensitive, calcium-release channels) associated with the sarcoplasmic reticulum. This provides more calcium for binding the troponin-C, which enhances inotropy. Finally, PK-A can phosphorylate myosin light chains, which may contribute to the positive inotropic effect of beta-adrenoceptor stimulation. Because there is generally some level of sympathetic tone on the heart, beta-blockers are able to reduce sympathetic influences that normally stimulate chronotropy (heart rate), inotropy (contractility), dromotropy (electrical conduction) and lusitropy (relaxation). Therefore, beta-blockers cause decreases in heart rate, contractility, conduction velocity, and relaxation rate. These drugs have an even greater effect when there is elevated sympathetic activity.
, inotropy (contractility), dromotropy (electrical conduction) and lusitropy (relaxation). Therefore, beta-blockers cause decreases in heart rate, contractility, conduction velocity, and relaxation rate. These drugs have an even greater effect when there is elevated sympathetic activity.")
25
Molecular Mechanism of Dipyridamol
Dipyridamole works by blocking the action of an enzyme found in platelets called phosphodiesterase. Inside the platelets phosphodiesterase normally breaks down a chemical called cyclic AMP. Dipyridamole causes the levels of cyclic AMP in the platelets to rise, because it stops phosphodiesterase from breaking it down. It inhibits the cellular reuptake of adenosine into platelets, red blood cells and endothelial cells leading to increased extracellular concentrations of adenosine. It also inhibits the enzyme adenosine deaminase, which normally breaks down adenosine into inosine. This inhibition leads to further increased levels of extracellular adenosine. Dipyridamole also inhibits the phosphodiesterase enzymes that normally break down cAMP (increasing cellular cAMP levels and blocking the platelet response to ADP) and/or cGMP (resulting in added benefit when given together with nitric oxide [NO] or statins).
 and/or cGMP (resulting in added benefit when given together with nitric oxide [NO] or statins).")
26
Hyperlipidemia Hyperlipidemia, hyperlipoproteinemia, or hyperlipidaemia involves abnormally elevated levels of any or all lipids and/or lipoproteins in the blood. It is the most common form of dyslipidemia (which also includes any decreased lipid levels). Lipids (fat-soluble molecules) are transported in a protein capsule. The size of that capsule, or lipoprotein, determines its density. The lipoprotein density and type of apolipoproteins it contains determines the fate of the particle and its influence on metabolism. Hyperlipidemias are divided in primary and secondary subtypes. Primary hyperlipidemia is usually due to genetic causes (such as a mutation in a receptor protein), while secondary hyperlipidemia arises due to other underlying causes such as diabetes. Lipid and lipoprotein abnormalities are common in the general population, and are regarded as a modifiable risk factor for cardiovascular disease due to their influence on atherosclerosis. In addition, some forms may predispose to acute pancreatitis.
. Lipids (fat-soluble molecules) are transported in a protein capsule. The size of that capsule, or lipoprotein, determines its density. The lipoprotein density and type of apolipoproteins it contains determines the fate of the particle and its influence on metabolism. Hyperlipidemias are divided in primary and secondary subtypes. Primary hyperlipidemia is usually due to genetic causes (such as a mutation in a receptor protein), while secondary hyperlipidemia arises due to other underlying causes such as diabetes. Lipid and lipoprotein abnormalities are common in the general population, and are regarded as a modifiable risk factor for cardiovascular disease due to their influence on atherosclerosis. In addition, some forms may predispose to acute pancreatitis.")
27
More than 650,000 people die every year of coronary heart disease (CHD) in the US alone. In 1984 it was demonstrated for the first time that there exists a link between serum cholesterol levels and risk to CHD. A 1% drop in serum cholesterol reduces the risk for CHD by 2%. Positive risk factors for CHD include Age (men>45 yrs, women>55 yrs); family history of premature CHD; smoking; hypertension (>140/90 mm Hg); low HDL cholesterol (<35 mg/dl); obesity (>30% overweight); diabetes; and high LDL (>160 mg/dl). Negative risk factors include high HDL levels (>60 mg/dl). Cholesterol does not occur alone in plasma. It is always associated with lipoproteins. Lipoproteins are proteins carrying lipids. Cholesterol is one of the lipids. Long chain fatty acids are also carried by these lipoproteins in the form of triglycerides (TG). Lipoproteins are actually aggregates. There are several forms of lipoproteins, very low density lipoproteins (VLDL), intermediate density lipoproteins (IDL), low density lipoproteins (LDL), and high density lipoproteins (HDL), depending on the density of their packing or alternatively their size. The term hyperlipidemia refers to the excessive lipid content in the blood plasma. A lipid profile of patient's blood plasma is the distribution in concentration of various forms lipoproteins.
. Lipoproteins are actually aggregates. There are several forms of lipoproteins, very low density lipoproteins (VLDL), intermediate density lipoproteins (IDL), low density lipoproteins (LDL), and high density lipoproteins (HDL), depending on the density of their packing or alternatively their size. The term hyperlipidemia refers to the excessive lipid content in the blood plasma. A lipid profile of patient s blood plasma is the distribution in concentration of various forms lipoproteins.")
28
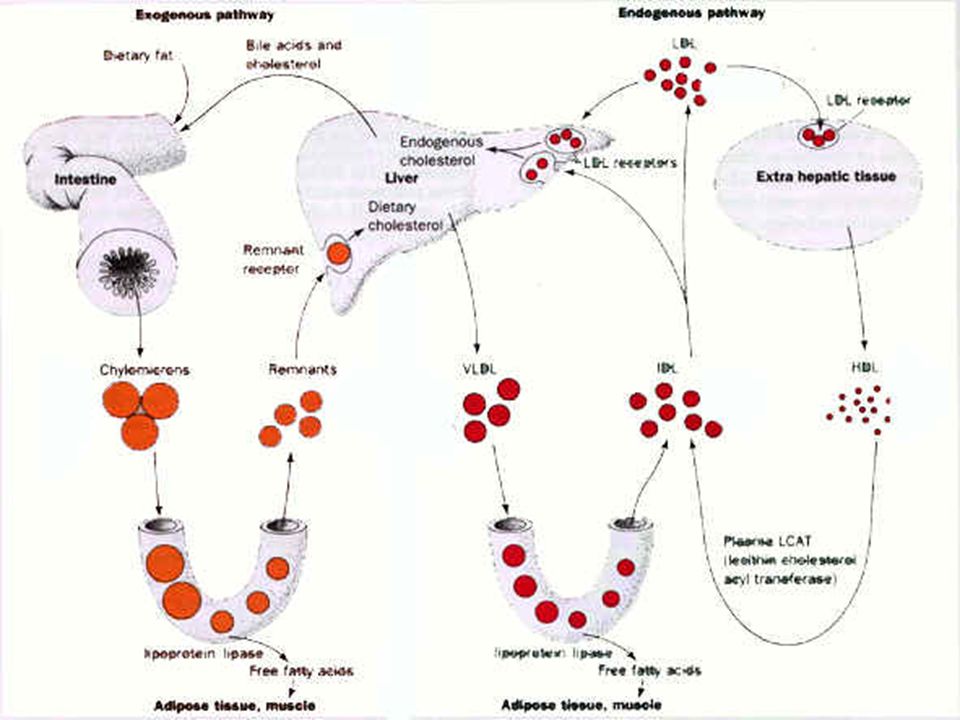
Mechanism of Lipid Transport
Dietary fat including cholesterol and triglycerides are absorbed in the intestine and released in the blood stream as chylomicrons. These are least dense particles having very high proportion of triacylglycerides. Lipoprotein lipase acts on these particles to release some free fatty acids that deposit in adipose tissues. The remnants of chylomicrons are picked up by the liver which has a receptor specific to chylomicron remnants. After further clean up liver releases particles called the very low density lipoproteins in the blood. These have lower triacyl glycerides than chylomicrons. Once again LPL works on these VLDL particles releasing more free fatty acids and changing the content of the particles to IDL and LDL. There are LDL receptors on the cell membranes of the extrahepatic cells which can pick up the LDL particles. This is how cholesterol reaches the interior of normal cells. Within cells, LDL particles are repackaged. Excess cholesterol is esterified and stored. Excess cholesterol suppresses the biosynthesis of LDL-receptors so that intake of cholesterol decreases. It also suppresses cholesterol biosynthesis. Repackaged LDL particles called HDL particles are then released into the blood stream. These particles are sensed by the liver through the HDL-receptors. Thus the liver gets constant information as to how much LDL and HDL are present in the blood.

30
Strategies for treating Hyperlipidemias

31
Statins inhibit the rate-limiting step in the biosynthesis of cholesterol - HMG -CoA reductase. Decreased cholesterol biosynthesis steps up the levels of the LDL-receptor resulting in the positive cycle for lowered cholesterol levels in serum. For patients who have familial hypercholesterolemia due to defective LDL-receptor genes these drugs are not effective. Statins are most effective cholesterol lowering drugs. Statins lower total cholesterol and LDL particles. These are competitive inhibitors. The HMG-CoA has a conformation similar to the lactone moiety of statins resulting in binding at the same site without any productive effect. All statins are highly protein bound (95-98%) except for pravastatin (50%, due to carboxylate moiety). Most statins have a short half-life of about 1-3 hr except for atorvastatin which has a t1/2 of about 14 h. A point drop in LDL could be reasonably expected with most statins after a therapy of about 1 month. A combination therapy (with bile acid sequestering agents) is helpful for particularly difficult cases. Although possible, statins typically do not affect the concentrations of steroid hormones in circulation.
 except for pravastatin (50%, due to carboxylate moiety). Most statins have a short half-life of about 1-3 hr except for atorvastatin which has a t1/2 of about 14 h. A point drop in LDL could be reasonably expected with most statins after a therapy of about 1 month. A combination therapy (with bile acid sequestering agents) is helpful for particularly difficult cases. Although possible, statins typically do not affect the concentrations of steroid hormones in circulation.")
32
Hypertension

33
Hypertension & Blood Pressure
Hypertension is a condition in which the blood pressure is persistently higher than normal Measurement is indirect Blood pressure is silent Hypertensive crisis: acute, life threatening rise in blood pressure associated with acute end-organ damage.

34
Risk Stratification Hypertension Heart Smoking
Major Cardiovascular Risk Factors Hypertension Smoking Obesity (BMI > 30) Physical inactivity Dyslipidemia Diabetes mellitus Microalbuminuria or GFR < 60ml/min Advanced age Men > 55, women > 65 Family history of premature CV disease Target Organ Disease Heart Left ventricular hypertrophy CAD Angina and/or prior MI Prior coronary revascularization Heart failure Brain Stroke or TIA Chronic renal insufficiency Peripheral arterial disease Retinopathy
 Physical inactivity. Dyslipidemia. Diabetes mellitus. Microalbuminuria or GFR < 60ml/min. Advanced age. Men > 55, women > 65. Family history of premature CV disease. Target Organ Disease. Heart. Left ventricular hypertrophy. CAD. Angina and/or prior MI. Prior coronary revascularization. Heart failure. Brain. Stroke or TIA. Chronic renal insufficiency. Peripheral arterial disease. Retinopathy.")
35
Therapeutic Treatment Options
Diuretics Beta blockers ACE inhibitors Angiotensin II receptor blockers Calcium channel blockers Alpha blockers Centrally acting alpha agonists Direct vasodilators Peripheral adrenergic blockers

36
Therapeutic Options: Beta Blockers Inhibit sympathetic stimulation on Beta-1 receptors heart Beta-2 receptors blood vessels, lungs Cardioselective vs. Nonselective Intrinsic sympathomimetic activity (ISA)
.")
37
Antihypertensive Agents
Hypertension (HTN) - An inc. in BP such that systolic is > 140 mm/hg & diastolic > 90 mm/hg on 2 or more occasions after initial screening * Exact Origin - unknown. Contributing Factors - family hx, hyperlipidemia, African American background, diabetes, obesity, aging, stress, excessive ETOH & smoking. Secondary HTN is about 10% of HTN, related to endocrine or renal disorders
 - An inc. in BP such that systolic is > 140 mm/hg & diastolic > 90 mm/hg on 2 or more occasions after initial screening. * Exact Origin - unknown. Contributing Factors - family hx, hyperlipidemia, African American background, diabetes, obesity, aging, stress, excessive ETOH & smoking. Secondary HTN is about 10% of HTN, related to endocrine or renal disorders.")
38
Renin-angiotensin system
Kidneys and blood vessels strive to regulate and maintain a “normal” BP. The kidneys regulate blood pressure via the renin-angiotensin system. Renin (from the renal cells) stimulates production of angiotensin I & then AT- II (a potent vasoconstrictor), causes the release of aldosterone (adrenal hormone that promotes sodium retention and then water retention). Retention of sodium and water causes fluid volume to increase, thus elevating blood pressure. N.E. , an adrenal hormone of the sympathetic nervous system, increases blood pressure.
 stimulates production of angiotensin I & then AT- II (a potent vasoconstrictor), causes the release of aldosterone (adrenal hormone that promotes sodium retention and then water retention). Retention of sodium and water causes fluid volume to increase, thus elevating blood pressure. N.E. , an adrenal hormone of the sympathetic nervous system, increases blood pressure.")
39
CARE APPROACH Step 1 Diuretic, Beta Blocker, Calcium blocker, Angiotensin-converting enzyme Step Diuretic with beta blocker Sympatholytics Step Direct-acting vasodilator Sympatholytic with diuretic Step Adrenergic neuron blocker Combinations from steps I, II & III

40
Antihypertensive Agents
Diuretics - * Promote Na depletion dec. in extra cellular fluid (ECF) * First line drug for mild HTN * Hydrochlorothiazide most frequently prescribed for first line drug of mild HTN * Can be used alone or w/ other antiHTN agents
 * First line drug for mild HTN. * Hydrochlorothiazide most frequently prescribed for first line drug of mild HTN. * Can be used alone or w/ other antiHTN agents.")
41
ANTIHYPERTENSIVE AGENTS
SYMPATHOLYTICS (SYMPATHETIC DEPRESSANTS) 1. BETA-ADRENERGIC 2. CENTRAL ACTING SYMPATHOLYTICS 3. ALPHA-ADRENERGICS 4. ADRENERGIC NEURON BLOCKERS 5. ALPHA & BETA ADRENERGIC BLOCKERS
 1. BETA-ADRENERGIC. 2. CENTRAL ACTING SYMPATHOLYTICS. 3. ALPHA-ADRENERGICS. 4. ADRENERGIC NEURON BLOCKERS. 5. ALPHA & BETA ADRENERGIC BLOCKERS.")
42
Beta-Adrenergic Blockers (Beta Blockers)
Atenolol (Tenormin), Metoprolol (Lopressor) - Beta-1 cardio selective Nadolol (Corgard), Propranolol (Inderal) - Nonselective Beta-1, Beta-2 - May be combined w/ a diuretic - Reduces cardiac output (CO) by diminishing sympathetic nervous system response
, Metoprolol (Lopressor) - Beta-1 cardio selective. Nadolol (Corgard), Propranolol (Inderal) - Nonselective Beta-1, Beta-2. - May be combined w/ a diuretic. - Reduces cardiac output (CO) by diminishing sympathetic nervous system response.")
43
- With continued use the vascular resistance diminished & BP lowered
- Reduces HR & contractility - Reduces renin release from kidneys Nonselective = inhibits Beta-1 (heart) & Beta-2 (bronchial) receptors - HR slows & BP decreases - Bronchoconstriction occurs Cardio selective - Preferred - acts mainly on Beta-1 receptors & bronchospasms - not absolute protection *Use cautiously in patients w/ pulmonary history*
 & Beta-2 (bronchial) receptors. - HR slows & BP decreases. - Bronchoconstriction occurs. Cardio selective - Preferred - acts mainly on Beta-1 receptors & bronchospasms - not absolute protection. *Use cautiously in patients w/ pulmonary history*")
44
(2) Centrally Acting Sympatholytics (Adrenergic Blockers)
Clonidine HCL (Catapres), Methyldopa (Aldomet) - Stimulate Alpha-2 receptors dec. sympathetic activity dec. epi., norepi. & dec.renin release dec. peripheral vascular resistance - Can be used w/ other agents - Clonidine = a new transdermal preparation - provides a 7 day duration of action - Used w/ diuretics – to prevent NA+ and fluid retention
, Methyldopa (Aldomet) - Stimulate Alpha-2 receptors dec. sympathetic activity dec. epi., norepi. & dec.renin release dec. peripheral vascular resistance. - Can be used w/ other agents. - Clonidine = a new transdermal preparation - provides a 7 day duration of action. - Used w/ diuretics – to prevent NA+ and fluid retention.")
45
(3) Alpha - Adrenergic Blockers
Prazosin HCL (Minipress) - Blocks alpha adrenergic receptors vasodilatation & a dec. in BP - Helps maintain renal blood flow - Useful in patients with lipid abnormalities - decs. VLDL & LDL - responsible for build-up of fatty plaques in arteries & incs. HDL. - Can cause Na & H2O retention - diuretics may be added
 - Blocks alpha adrenergic receptors vasodilatation & a dec. in BP. - Helps maintain renal blood flow. - Useful in patients with lipid abnormalities - decs. VLDL & LDL - responsible for build-up of fatty plaques in arteries & incs. HDL. - Can cause Na & H2O retention - diuretics may be added.")
46
Safe for diabetics, do not affect respiratory function.
Used in HTN, refractory CHF, Benign prostatic hypertrophy (BPH) Side effects – dizziness, drowsiness, impotence, vertigo, urinary frequency, tinnitus, dry mouth Adverse - Orthostatic hypotension, palpitations, tachycardia When taken with ETOH or other antihyper. severe hypotension
 Side effects – dizziness, drowsiness, impotence, vertigo, urinary frequency, tinnitus, dry mouth. Adverse - Orthostatic hypotension, palpitations, tachycardia. When taken with ETOH or other antihyper. severe hypotension.")
47
(4) Adrenergic Neuron Blockers (Peripherally acting sympatholytics)
* Potent drugs that block norepi. form sympathetic nerve endings a dec. in norepi. dec. in BP * Decrease in both cardiac output & peripheral vascular resistance Reserpine (Serpasil) & guanethidine (Ismelin) - Potent - used for severe HTN * Step IV drugs - alone or with diuretics to dec. peri. edema * Common SE = Orthostatic Hypotension*
 & guanethidine (Ismelin) - Potent - used for severe HTN. * Step IV drugs - alone or with diuretics to dec. peri. edema. * Common SE = Orthostatic Hypotension*")
48
(5) Alpha-1 & Beta-1 Adrenergic blockers
Carteolol (Cartrol), Labetalol (Trandate) - Blocks both alpha-1 & beta-1 receptors - Block alpha-1 = dilation of arterioles & veins -Effect on alpha receptors stronger than on beta receptors so have a dec. BP & pulse rate - Block beta-1 lead to decreased HR & AV contractility - Large doses could block beta-2 receptors inc. in air way resistance - Do not give to severe asthmatics. AV block Side Effects = Orthostatic Hypotension, GI, nervousness, dry mouth & fatigue
, Labetalol (Trandate) - Blocks both alpha-1 & beta-1 receptors. - Block alpha-1 = dilation of arterioles & veins. -Effect on alpha receptors stronger than on beta receptors so have a dec. BP & pulse rate. - Block beta-1 lead to decreased HR & AV contractility. - Large doses could block beta-2 receptors inc. in air way resistance - Do not give to severe asthmatics. AV block. Side Effects = Orthostatic Hypotension, GI, nervousness, dry mouth & fatigue.")
49
Direct - Acting Arteriolar Vasodilators - potent
Hydralazine (Apresoline) - Mod. to severe HTN Sodium Nitroprusside (Nipride) - Very potent - for hypertensive Emergencies - Act by relaxing smooth muscles of bld. vessels - mainly arteries vasodilation - Inc. blood flow to brain & kidneys - With vasodilation the BP dec., Na & H2O retained peripheral edema. Diuretics used to counter this SE - Side Effects = numerous - tachycardia, palpitations, edema, dizzy, GI bleeding
 - Mod. to severe HTN. Sodium Nitroprusside (Nipride) - Very potent - for hypertensive Emergencies. - Act by relaxing smooth muscles of bld. vessels - mainly arteries vasodilation - Inc. blood flow to brain & kidneys. - With vasodilation the BP dec., Na & H2O retained. peripheral edema. Diuretics used to counter this SE. - Side Effects = numerous - tachycardia, palpitations, edema, dizzy, GI bleeding.")
50
Captopril (Capoten), Enalapril (Vasotec), Lisinopril (Zestril)
Angiotensin Antagonists - Angiotensin-Converting Enzyme Inhibitors (ACE inhibitors) Captopril (Capoten), Enalapril (Vasotec), Lisinopril (Zestril) - Prevents conversion of Angiotensin I to angiotensin II (vasoconstrictor) & blocks release of aldosterone. Aldosterone promotes Na retention & K excretion. Block aldosterone & Na excreted, but H2O & K retained - Used to treat HTN primarily, - but not a 1st line drug. Also used in heart failure. - SE = hyperkalemia & 1st dose hypotension (more common with comb. Diuretic & ACE inhibitor.
 Captopril (Capoten), Enalapril (Vasotec), Lisinopril (Zestril) - Prevents conversion of Angiotensin I to angiotensin II (vasoconstrictor) & blocks release of aldosterone. Aldosterone promotes Na retention & K excretion. Block aldosterone & Na excreted, but H2O & K retained. - Used to treat HTN primarily, - but not a 1st line drug. Also used in heart failure. - SE = hyperkalemia & 1st dose hypotension (more common with comb. Diuretic & ACE inhibitor.")
51
Angiotensin II receptor Antagonists (Blockers) - A - II Blockers
Losartan (Cozaar) - Newer drugs similar to ACE inhibitors + prevent release of aldosterone (Na+ retaining hormone) - Act on renin - angiotensin system - Diff between ACE &A II is A-II blockers block angiotensin from angiotensin I receptors found in many tissues - blocks at receptor site. - A-II blockers cause vasodilation & dec. peripheral resistance
 - Newer drugs similar to ACE inhibitors + prevent release of aldosterone (Na+ retaining hormone) - Act on renin - angiotensin system. - Diff between ACE &A II is A-II blockers block angiotensin from angiotensin I receptors found in many tissues - blocks at receptor site. - A-II blockers cause vasodilation & dec. peripheral resistance.")
52
ACE inhibitors inhibit the enzyme necessary for the conversion of A-I to A-II
A-II blockers - block angiotensin II from receptors in blood vessels, adrenals, and all other tissues.

53
Calcium Channel Blockers
Verapamil (Calan), Nifedipine (Procardia), Diltiazem (Cardizem) - Free calcium muscle contractility, peripheral resistance & BP So, Calcium blockers - Dec. calcium levels & promote vasodilation - Drugs can be used w/ clients prone to asthma - SE. Flushing, HA, dizziness, ankle edema, bradycardia, AV node block,
, Nifedipine (Procardia), Diltiazem (Cardizem) - Free calcium muscle contractility, peripheral resistance & BP . So, Calcium blockers. - Dec. calcium levels & promote vasodilation. - Drugs can be used w/ clients prone to asthma. - SE. Flushing, HA, dizziness, ankle edema, bradycardia, AV node block,")
Similar presentations