Download presentation
Presentation is loading. Please wait.

1
Bone Marrow Failure/ Aplastic Anemia
Dr. MERVAT A.HESHAM 2008

2
What is Aplastic Anemia?
Aplastic Anemia is a bone marrow failure disease. Bone marrow is a Factory of Blood Cells Red Blood Cell Platelets White Blood Cell Help to save a Life

3
Aplastic Anemia patients
Aplastic Anemia patients have decreased amounts of: - Red Blood Cells - White Blood Cells - Platelets Help to save a Life

4
Functions of Blood Cells
Red Blood Cells Carry oxygen to all body organs White Blood Cells Fight infection and keep you healthy Platelets Help control bleeding Help to save a Life

5
Help to save a Life www.aaaoi.org
Symptoms Low Red Blood Cell Fatigue, Headache, Inability to Concentrate Low White Blood Cell Viral Infections, Bacterial Infections Low Platelets Easy Bruising, Nosebleeds, Petichiae Help to save a Life

6
DEFINITION A disorder of the hemtopoietic system characterized by:
Bone marrow - marked reduction of all 3 cell lines Peripheral blood - pancytopenia

7
PATHOGENESIS Stem cell failure resulting from:
1-An acquired intrinsic stem cell defect 2-An environmental cause Immune mechanisms Growth factor deficiency Defects in the microenvironment

8
Epidemiology Incidence: 5-10:106 per year Age: 15 –30 years
Sex: M = F Aplastic anemia

9
Etiology Hereditary Acquired 1-Schwacman – Diamond
2-Fanconi’s anemia syndrome 3-Dyskeratosis congenita Acquired 1-Idiopathic 2- Drugs: dose related idiosyncratic 3-Radiation 4-Chemicals 5-Viruses 6-Pregnancy 7-PNH 8-Disorders of immune system

10
Clinical manifestations
Insidious onset Manifestations caused by pancytopenia Anemia - weakness, fatigue Thrombocytopenia – bleeding Neutropenia - infections

11
Diagnosis Peripheral blood Bone marrow biopsy *Pancytopenia
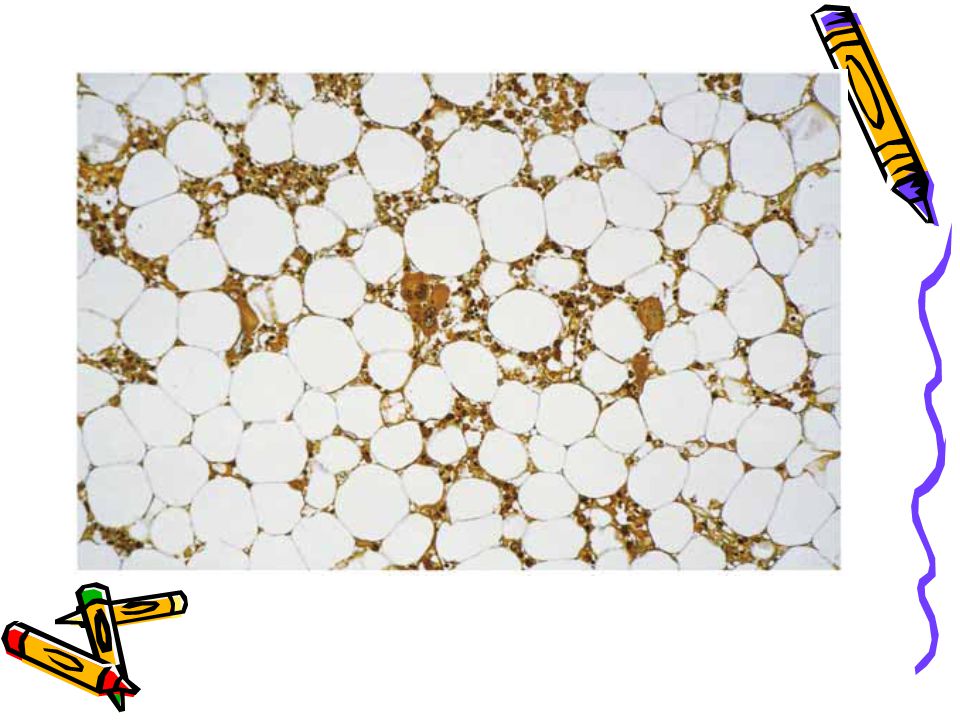
*Normocytic-normochromic anemia *Low reticulocyte index Bone marrow biopsy *Empty fatty spaces *Few hematopoietic cells *Lymphocytes and plasma cells

12
Bone Marrow Failure Congenital/ Syndromic Acquired

13
Acquired Aplastic Anemia

14
Acquired Aplastic Anemia
**Secondary **Idiopathic

15
Secondary AA 1-Meds/ toxins 2- Radiation
Chemo Chloramphenicol, benzene, carbamazapine, indomethacin, cimetidine, sulfas, acetazolamide, lithium 2- Radiation 3-Viruses - EBV, HIV, parvo, hepatitis

16
4-Paroxysmal Nocturnal Hemoglobinurea
5-Malnutrition 6-Myelodysplastic syndromes 7-Thymoma

17
PATHOPHYSIOLOGY Direct toxic injury to hematopoietic stem cells can be induced by exposure to ionizing radiation, cytotoxic chemotherapy, or benzene. These agents can crosslink DNA and induce DNA strand breaks leading to inhibition of DNA and RNA synthesis.

18
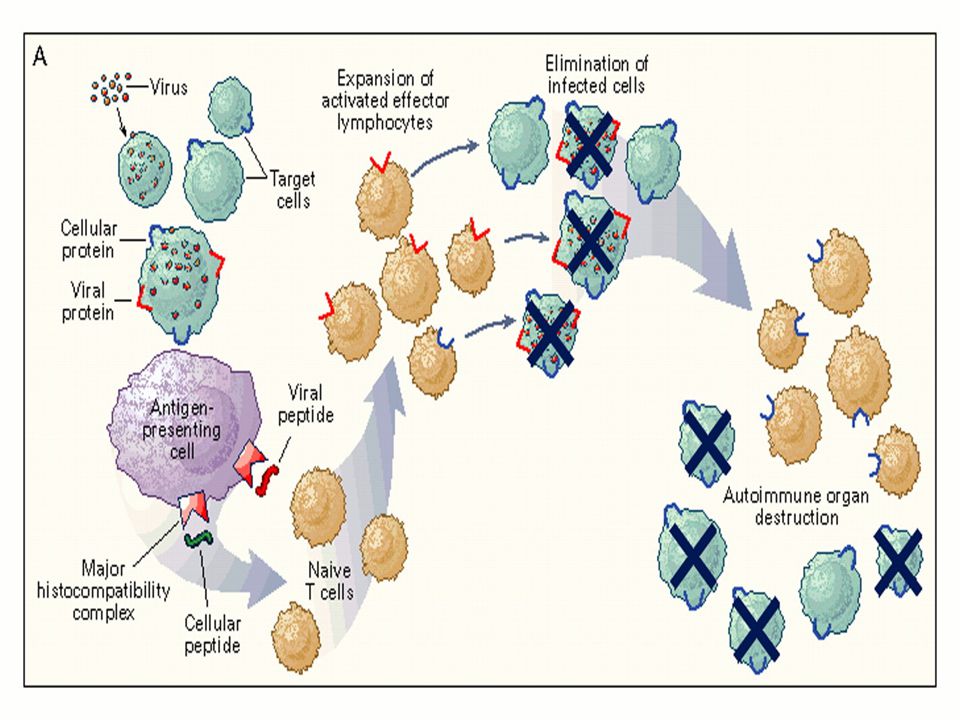
2-Immune-mediated destruction of hematopoietic stem cells
-- Direct killing of the stem cells has been hypothesized to occur via interations between Fas ligand expressed on the T-cells and Fas (CD95) present on the stem cells, which triggers programmed cell death (apoptosis). -- T-lymphocytes also may suppress stem cell proliferation by elaborating soluble factors including interferon-γ.
 present on the stem cells, which triggers programmed cell death (apoptosis). -- T-lymphocytes also may suppress stem cell proliferation by elaborating soluble factors including interferon-γ.")
19
-T cells from aplastic anemia patients secrete IFN-ã and tumor necrosis factor (TNF).
-IFN-ã and TNF are potent inhibitors of both early and late hematopoietic progenitor cells . -Both of these cytokines suppress hematopoiesis by their effects on the mitotic cycle and, more importantly, by the mechanism of cell killing. -Activation of the Fas receptor on the hematopoietic stem cell by the Fas ligand present on the lymphocytes leads to apoptosis of the targeted hematopoietic progenitor cells.

20
*Cytotoxic T cells also secrete interleukin-2 (IL-2), which causes polyclonal expansion of the T cells. * IFN-ã also induces the production of the toxic gas nitric oxide, diffusion of which causes additional toxic effects on the hematopoietic progenitor cells.

21
Suppress proliferation
with ligand, signals apoptosis Young NEJM 1997

23
Idiopathic AA *70% or more of cases Higher in SE Asia M = F

24
AA - Clinical ** Symptoms are due to pancytopenia:
pallor, mucosal bleeding, ecchymoses, or petechiae and bacterial or fungal infections.. ** Hepatosplenomegaly and lymphadenopathy do not occur; their presence suggests an underlying leukemia. ** Hyperplastic gingivitis is also a symptom of aplastic anemia.

25
AA - Labs No RBC = pale, tachycardic No plt = bruising, bleeding
No WBC = infection Retic < 1% Plt < 20,000 ANC < 500

26
AA - Labs Marrow : < 25% cellularity

28
AA - Evaluation *CBC w/ diff and retic *Send DEB (Fanconi’s test)
*Bone marrow *Send DEB (Fanconi’s test) *Send Hep A, B, C, D titers HIV *Test for PNH (CD55, CD59) *HLA typing *Fetal hemoglobin *Liver and renal function chemistries
 *Send Hep A, B, C, D titers HIV. *Test for PNH (CD55, CD59) *HLA typing. *Fetal hemoglobin. *Liver and renal function chemistries.")
29
*Quantitative immunoglobulins, C3, C4, and complement.
* Autoimmune disease evaluation: Antinuclear antibody (ANA), total hemolytic complement (CH50), Coombs’ test. * HLA typing: Patient and family done at the time of diagnosis of severe aplastic anemia to ensure a timely transplant.
, total hemolytic complement (CH50), Coombs’ test. * HLA typing: Patient and family done at the time of diagnosis of severe aplastic anemia to ensure a timely transplant.")
30
CLASSIFICATION Designation Criteria Peripheral blood BM biopsy
Severe aplastic anemia -2 / 3 values- Neutrophils < 500/mL -Platelets < 20,000/ ul- -Reticulocyte index < 1% -Marked hypocellular < 25% cellularity -Moderate hypocellular <25-50% -normal cellularity with <30% of remaining cell hematopoietic Very severe aplastic anemia As above but neutrophils < 200/mL Infection present

31
Help to save a Life www.aaaoi.org
Treatment Options Bone Marrow Transplant Growth Hormones Immune Suppressive Therapy Supportive Care Help to save a Life

32
TREATMENT 1-Withdrawal of the etiologic agent 2-Supportive treatment
Blood and platelet transfusion used with caution- sensitization (filtered) 3-Allogeneic BMT -Preferably from sibling -Curative in 60-90% of patients -Applicable only for a third of patients *Immunosuppression Cyclosporin + ATG Corticosteroids High dose cyclophosphamide *G-CSF/ GM-CSF/ EPO - maybe **Response rate 50-70% Occurs 2-3 months post Rx.
 3-Allogeneic BMT. -Preferably from sibling. -Curative in 60-90% of patients. -Applicable only for a third of patients. *Immunosuppression. Cyclosporin + ATG. Corticosteroids. High dose cyclophosphamide. *G-CSF/ GM-CSF/ EPO - maybe. **Response rate 50-70% Occurs 2-3 months post Rx.")
33
AA Newer *Mycophenolate mofetil (MMF) - cytotoxic to T cells
*Monoclonal Ab against IL-2 receptor which is present on activated lymphocytes

34
AA - Outcomes Age, Younger is better BMT Immunosuppression - 60 - 80%
< 20 yr with a sib… 75% yr with a sib…60% < 20 yr unrelated BMT… 40% yr unrelated BMT…35% Immunosuppression % But for how long and consequences…

35
Fanconi Anemia

36
History: Guido Fanconi
Fanconi Anemia (Fanconi pancytopenia syndrome): brothers with pancytopenia and physical abnormalities, “perniziosiforme” Fanconi Syndrome (renal Fanconi syndrome): 1936 – Ricketts, growth retardation, proteinuria, glucosuria, and proximal renal tubular acidosis Alter, FA101 (2006)
: brothers with pancytopenia and physical abnormalities, perniziosiforme Fanconi Syndrome (renal Fanconi syndrome): 1936 – Ricketts, growth retardation, proteinuria, glucosuria, and proximal renal tubular acidosis. Alter, FA101 (2006)")
37
Fanconi Anemia (FA) Rare (< 1/ 100,000 births) Autosomal recessive
Many physical features But up to 20-25% will have no physical findings Mean age at dx 7.8 yrs

38
Autosomal Recessive Inheritance

39
FA- Clinical Abnormality % of FA Patients Skin 60% Short Stature 57%
Upper Limb Abnl 48% Head/ Microcephaly 27% Renal 23% Dev. Delay 13% None Reported 20% Short Stature Only 1% Skin Only 3%

42
Clinical Features Progressive bone marrow failure
Most common etiology of inherited bone marrow failure Others include dykeratosis congenita, amegakaryocytic thrombocytopenia, Schwachman-Diamond syndrome Increased risk of MDS and AML (15,000x) Many have monosomy 7, or duplication of 1q (Auerbach et al., Cancer Genet Cytogenet 1991)
 Many have monosomy 7, or duplication of 1q (Auerbach et al., Cancer Genet Cytogenet 1991)")
43
Clinical Features Increased risk of solid tumor formation (hepatic, esophageal, oropharyngeal, vulvar) Average age at diagnosis is 23* Cumulative incidence ~30% by age 45** *Shimamura et al., Gene Reviews 2002 (genetests.org) **Alter et al. Blood 2003
 **Alter et al. Blood")
44
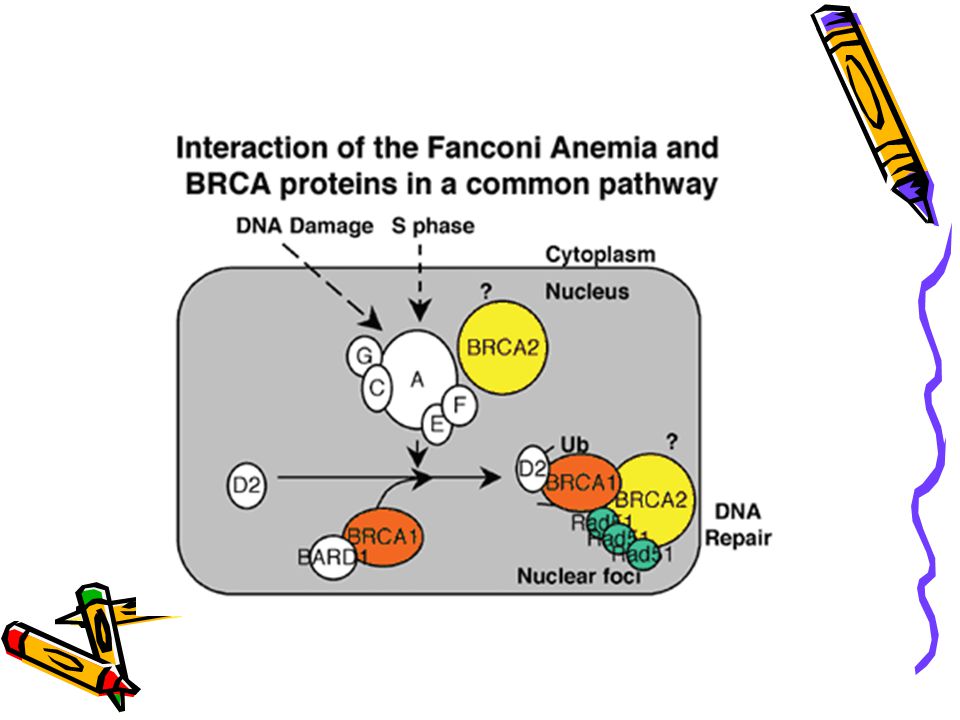
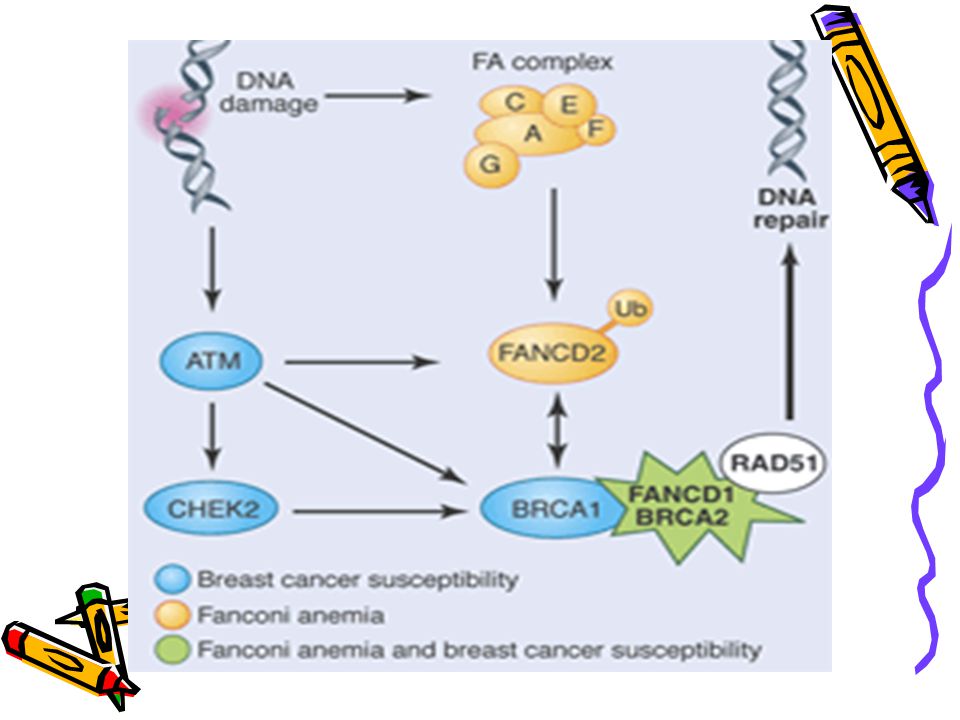
FA - genetics Identification of subtypes (compliment groups)
A, B, C, D1, D2, E, F, G Identical clinically Sub-units of a common protein/ common pathway Protein modifies FANCD2 FANCD2 interacts with BRCA1 and 2 BRCA1 and 2 needed for DNA repair

47
PATHOPHYSIOLOGY DNA damage activates a complex consisting of Fanconi proteins A, C, G, and F. This in turn leads to the modification of the FANCD2 protein. This protein interacts, for example, with the breast cancer susceptibility gene BRCA1.

48
*Fanconi anemia cells are characterized by hypersensitivity to chromosomal breakage as well as hypersensitivity to G2/M cell cycle arrest induced by DNA cross-linking agents. *In addition there is sensitivity to oxygen-free radicals and to ionizing radiation.

49
Diagnosis -*Pts. with congenital abnormalities are often diagnosed as neonates/infants *Others may be diagnosed when hematological problems occur *Median age of onset of pancytopenia is 7 Usually normal CBC at birth *First develop macrocytosis, then thrombocytopenia, and eventually neutropenia

50
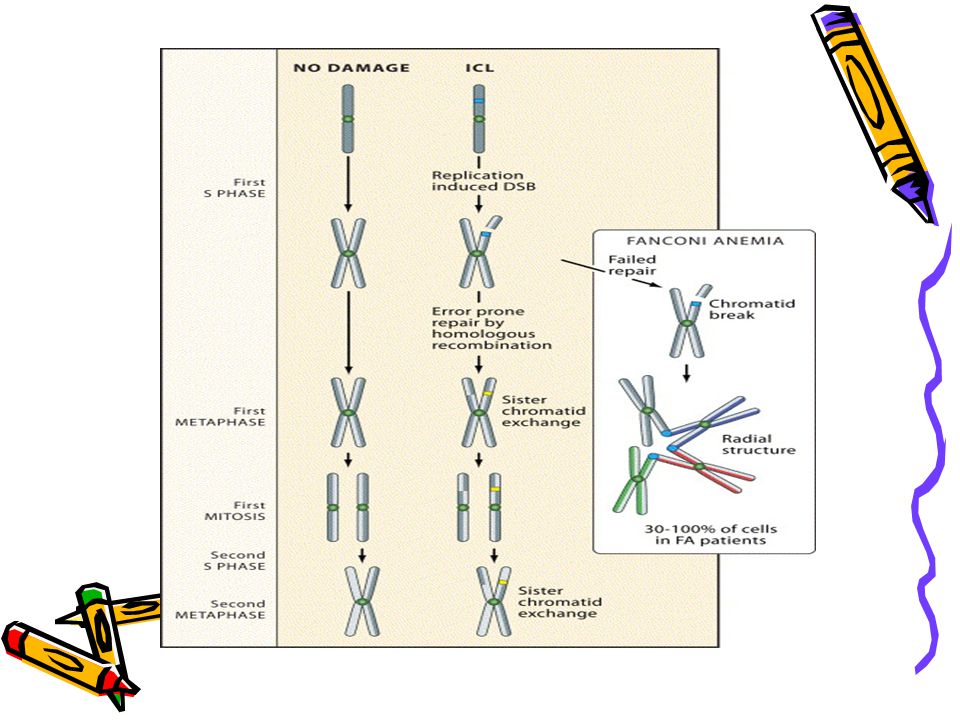
Diagnosis Based on chromosomal hypersensitivity to cross-linking agents Chromosome fragility test: Mitomycin C (MMC) or diepoxybutane (DEB) added to lymphoctyes – increases the number of chromosome breaks and radial structures Very specific for FA, regardless of severity of disease Can do chromosome breakage analysis on amniotic cells, chorionic villus cells or fetal blood
 or diepoxybutane (DEB) added to lymphoctyes – increases the number of chromosome breaks and radial structures. Very specific for FA, regardless of severity of disease. Can do chromosome breakage analysis on amniotic cells, chorionic villus cells or fetal blood.")
52
Chromosome breakage in Fanconi Anemia cells
FA cells were treated with mitomycin C and harvested in metaphase. Typical abnormalities include radial formation (green circle) and chromosome breaks (red arrows).
 and chromosome breaks (red arrows).")
53
Initial management Refer for genetic counseling
Testing of siblings Renal ultrasound, hearing test, eye exam Endocrine evaluation if evidence of growth failure (check growth hormone levels, TSH) Referral to hand surgeon for radial ray defects Bone marrow biopsy
 Referral to hand surgeon for radial ray defects. Bone marrow biopsy.")
54
Management Bone marrow failure Transfusions
Androgens (e.g. oral oxymethalone) – can improve blood counts in 50% of pts. Side effects: Masculinization, acne, hyperactivity, premature closure of epiphyses, liver toxicity, hepatic adenomas Growth factors (G-CSF, CM-CSF) – should not be used in patients with clonal cytogenetic abnormalities Bone marrow transplantation FA cells are very sensitive to radiation and alkylating agents – can use greatly reduced doses 2-yr. survival 70% for allo;* % for MUD** *Guardiola et al. Bone Marrow Transplant 1998; **MacMillan et al., Br J Haematol 2000
 – can improve blood counts in 50% of pts. Side effects: Masculinization, acne, hyperactivity, premature closure of epiphyses, liver toxicity, hepatic adenomas. Growth factors (G-CSF, CM-CSF) – should not be used in patients with clonal cytogenetic abnormalities. Bone marrow transplantation. FA cells are very sensitive to radiation and alkylating agents – can use greatly reduced doses. 2-yr. survival 70% for allo;* 20-40% for MUD** *Guardiola et al. Bone Marrow Transplant 1998; **MacMillan et al., Br J Haematol")
55
Management - Gene therapy
*Goal is to permanently correct hematological manifestations by transducing hematopoietic progenitor cells with a vector containing the deficient gene *Knockout mice with FANCC using retroviral vectors - phenotypic correction (Gush et al., Blood 2000) *Knockout mice with FANCA and FANCC using lentiviral vectors – more promising (integrates into the genome) (Galimi et al. Blood 2002)
 *Knockout mice with FANCA and FANCC using lentiviral vectors – more promising (integrates into the genome) (Galimi et al. Blood 2002)")
56
Other Congenital Marrow Failures
Dystkeratosis Congenita Rare Different modes of inheritance Ectodermal dysplasia 50% develop aplastic anemia in midteens Schwachman-Diamond Cartilage-Hair Hypoplasia Familial Marrow Dysfunction

57
Marrow Failure Pearson’s syndrome Seckel’s syndrome
Amegakaryocytic Thrombocytopenia Noonan’s syndrome

58
-Pure Red Cell Aplasia (Diamond-Backfan)
Marrow Failure Single Cytopenias -Pure Red Cell Aplasia (Diamond-Backfan) -Congenital Neutropenia (Kostmann’s) Thrombocytopenia with Absent Radii
 -Congenital Neutropenia (Kostmann’s) -Thrombocytopenia with Absent Radii.")
59
PURE RED CELL APLASIA

60
Definition A syndrome characterized by Normocytic normochromic anemia
Reticulocytopenia <1% BM erythroblasts < 0.5% Aplasia selective to erythroid cell line only !

61
Epidemiology Relatively uncommon
May affect any age group but predominantly of infancy and childhood M=F No ethnic predisposition Of autosomal dominant inheritance

62
Etiology & Pathogenesis
Congenital hypoplastic anemia (Diamond-Blackfan syndrome) Acquired PRCA Primary Secondary
 Acquired PRCA. Primary. Secondary.")
63
Acquired PRCA Primary Secondary Autoimmune Preleukemic Idiopathic
Thymoma Hematologic malignancies Solid tumors Infections Chronic hemolytic anemias Collagen vascular diseases Pregnancy Severe renal failure Severe nutritional deficiencies Drugs & chemicals Miscellaneous

64
PRIMUM NON NOCERE

65
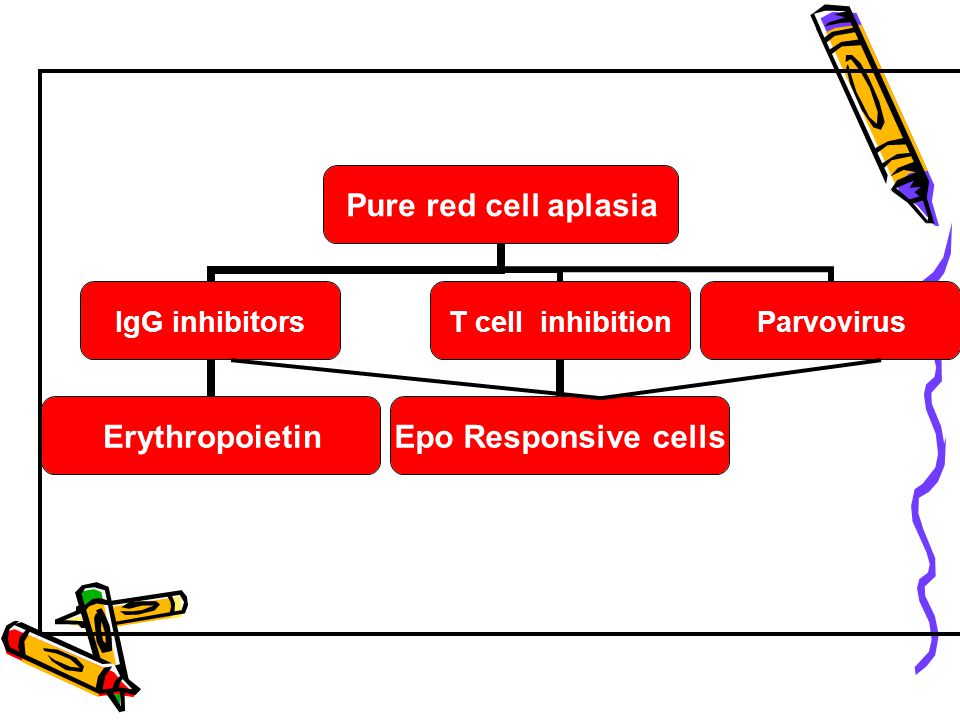
Mechanisms of Immunologic Inhibition
Antibodies directed against Erythropoietin Erythroblasts ? Cellular inhibition Inhibitory T cells NK cells

67
Clinical Manifestations
Symptoms of anemia *The median age at presentation of anemia is 2 months and the median age at diagnosis of DBA is 3 months. *Physical anomalies, excluding short stature *No hepatosplenomegaly. *Malignant potential In patients with long-standing PRCA – transfusional hemosiderosis

68
Laboratory Evaluation
Diagnostic criteria: --Normochromic, usually macrocytic anemia, relative to patient’s age and occasionally normocytic anemia developing in early childhood -- Reticulocytopenia -- Normal or only slightly decreased granulocyte count -- Normal or slightly increased platelet count Supportive criteria: -Typical physical abnormalities -Increased fetal hemoglobin -Increased erythrocyte adenosine deaminase (eADA) activity
 activity.")
69
BM *Absence of erythroblasts <1% on BM (absence of normoblasts, in some cases with relative increase in proerythroblasts or normal number of proerythroblasts with a maturation arrest). *normal myeloid and megakaryocytic series. *Usually – normal karyotype, except for preleukemic cases
. *normal myeloid and megakaryocytic series. *Usually – normal karyotype, except for preleukemic cases.")
71
Treatment Congenital Hypoplastic Anemia
Corticosteroids AlloBMT IL-3 –experimental Patients refractory to all treatments – regular transfusions & desferioxamine

72
Treatment Acquired PRCA
-Discontinuation of all drugs -R/O infections -If parvovirus suspected – high dose IgG -In the presence of thymoma – thymectomy

73
-In 30-40% erythropoiesis remits within 4-8 weeks
-Non-responding pts. – should be treated as primary acquired PRCA -Thymectomy in the absence of thymoma is not recommended -If an underlying disease – treat the disease

74
Treatment Acquired PRCA
For primary or secondary PRCA not responding to treatment of underlying disease: Prednisone Cyclophosphamide / azathioprine Cyclosporine ATG High dose IgG Plasmapheresis Splenectomy Rituximab

75
THANK YOU

Similar presentations






1 CHILDHOOD LEUKAEMIA. TA OGUNLESI (FWACP)2 LEUKAEMIA Heterogenous group of malignant disorders Characterised by uncontrolled clonal.>")










 Idiopathic Associated with other diseases (autoimmune, infectious, non-heme.>")



 Masood Anwar. Bone marrow failure syndromes can be defined as a group of diseases in which occurs failure on the part of bone marrow to produce.>")
 Course code: MLHE-201 Supervisor: Prof. Dr Magda Sultan Outcome : The student will know : -The causes and pathogenesis of.>")
