Download presentation
Presentation is loading. Please wait.

1
Rheumatology case ד"ר ענת שיימן

2
A 60-year-old man was evaluated in the rheumatology clinic because of a rash and worsening renal function. He had been well until three months earlier, when he began to have fatigue and weight gain, one week after returning from a visit to his son in Colorado. He could not fit into his shoes, and his wife noticed that he had facial swelling. Three days later, a rash appeared over his buttocks and feet.

3
The patient was a retired management consultant who lived in New Hampshire. He did not have photosensitivity, hair loss, Raynaud's phenomenon, keratoconjunctivitis sicca symptoms, or arthritis. An appendectomy had been performed two years earlier because of acute appendicitis. He drank approximately five alcoholic beverages per week and did not smoke cigarettes. He had no allergies. There was no family history of renal disease or autoimmune disorders. He was married, and his wife and a son and a daughter were well. He knew of no exposures to toxins or ill persons and did not recall tick bites. He had traveled recently only to Colorado and Connecticut.

4
He saw his primary care physician. The blood pressure was 154/82 mm Hg. There was 1+ bilateral pretibial and pedal edema. Tests for antineutrophil cytoplasmic antibodies and anti– streptolysin O were negative; a test for antinuclear antibodies was weakly positive (8 U; normal, less than 7.5 U).
..")
5
The results of chest radiography showed no abnormalities, except for blunting of the costophrenic angles that was thought to represent small effusions. An ultrasonographic study of the kidneys revealed hypoechoic areas in the left kidney and a normal-appearing right kidney. Furosemide and metolazone were started.

6
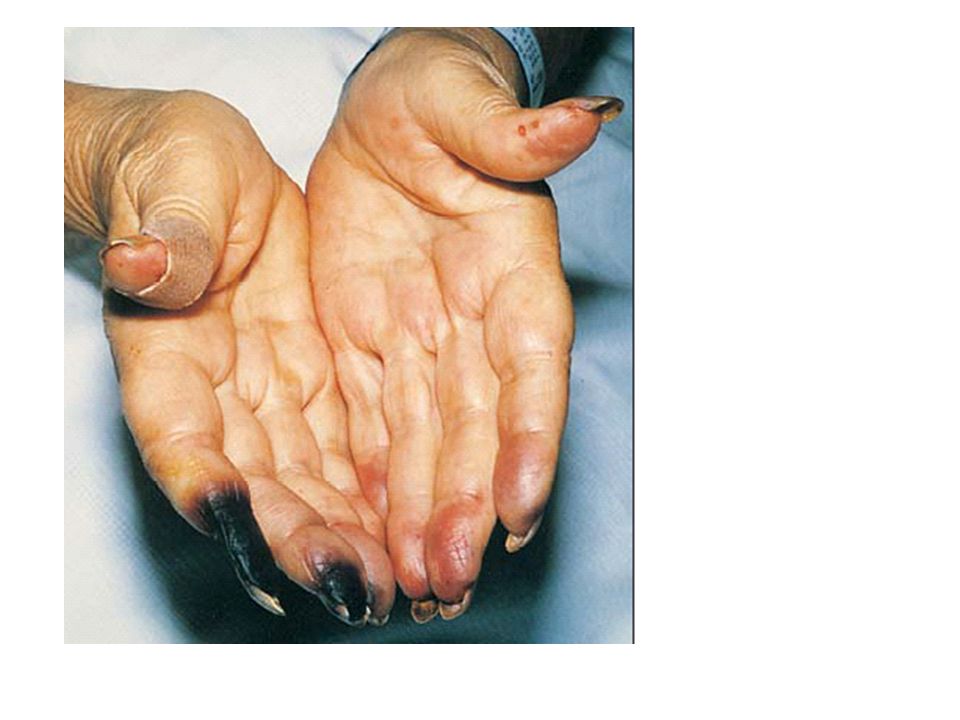
Despite increasing doses of furosemide, the patient continued to gain weight over the course of the next 10 days. He was referred to a nephrologist at another hospital. The blood pressure was 175/95 mm Hg, and he had gained more than 9 kg since the start of his illness. Palpable purpuric lesions were present on the buttocks and feet, including the soles.

7
Lab tests?

8
Serum creatinine level was 2.1 mg per deciliter (185.6 μmol per liter). Urinalysis revealed 3+ protein, 4+ blood, 10 to 15 red cells per high-power field, 5 to 10 white cells per high-power field, and many coarse and fine granular casts.

9
More tests?

10
Skin-biopsy specimen showed a leukocytoclastic vasculitis. Direct immunofluorescence staining of the biopsy specimen showed granular deposits consisting primarily of IgM and C3 in a vascular pattern in the papillary dermis, with very faint deposits of IgG and no IgA.

11
What should we do now?

12
A renal biopsy was performed one week later at that hospital, and a diagnosis of endocapillary proliferative glomerulonephritis, immune- complex type, was made. Immunofluorescence studies were reported to show mesangial and focal capillary-loop deposits of IgG, C3 (trace to 1+), and IgM (1+). Electron-microscopical examination revealed small mesangial and subendothelial electron- dense deposits. The findings were considered consistent with postinfectious glomerulonephritis, provided that other types of immune-complex glomerulonephritis could be ruled out — in particular, lupus nephritis.
, and IgM (1+). Electron-microscopical examination revealed small mesangial and subendothelial electron- dense deposits. The findings were considered consistent with postinfectious glomerulonephritis, provided that other types of immune-complex glomerulonephritis could be ruled out — in particular, lupus nephritis..")
13
Over the next two weeks, the patient continued to be fatigued, with a poor appetite; the serum creatinine level rose to a peak of 2.9 mg per deciliter (256.4 μmol per liter).
.")
14
What next?

15
Treatment with prednisone (60 mg per day), lisinopril, amlodipine, and omeprazole was started, and treatment with metolazone was discontinued. The patient was also still taking furosemide and atenolol. Over the next four weeks, the serum creatinine level fell to 1.9 mg per deciliter (167.9 μmol per liter), the patient's edema improved, the rash gradually resolved, and he felt more energetic. During the next week, however, the edema increased, and over the course of the next four weeks the creatinine level rose to 2.4 mg per deciliter (212.2 μmol per liter). A 24-hour urine collection yielded 5.7 g per deciliter of protein. Three weeks later, the patient was seen in the rheumatology clinic of this hospital.
, the patient s edema improved, the rash gradually resolved, and he felt more energetic. During the next week, however, the edema increased, and over the course of the next four weeks the creatinine level rose to 2.4 mg per deciliter (212.2 μmol per liter). A 24-hour urine collection yielded 5.7 g per deciliter of protein. Three weeks later, the patient was seen in the rheumatology clinic of this hospital..")
16
On examination He appeared chronically ill. Blood pressure 170/100 mm Hg, pulse 60, respiratory rate 14 breaths per minute. Cushingoid appearance, and there was diffuse anasarca. Marked (4+) pitting edema of both forearms and both legs, extending up to the thighs. Remainder of the physical and neurologic examinations were normal.
 pitting edema of both forearms and both legs, extending up to the thighs. Remainder of the physical and neurologic examinations were normal..")
17
Summery This 60-year-old man had a sudden onset of constitutional symptoms; leukocytoclastic cutaneous vasculitis; facial, arm, and leg edema; and hypertension, within two weeks after returning from a visit to Colorado. Laboratory evaluation revealed anemia and renal insufficiency, with microscopic hematuria and proteinuria in the nephrotic range. Serum complement levels were decreased. Testing for cryoglobulins and antineutrophil cytoplasmic antibodies had been negative.

18
Diagnosis?

19
Dr. Franklin D. Segall (Nephrology, Mount Auburn Hospital): When I first saw the patient, the urine sediment suggested glomerular inflammation. Because of the rash, I considered Henoch–Schönlein purpura and obtained a skin-biopsy specimen, which showed no IgA deposition. When the renal biopsy suggested lupus nephritis, I started the patient on steroid treatment; he had initial improvement, but his renal function continued to deteriorate, and I sent him to Dr. Kay.
: When I first saw the patient, the urine sediment suggested glomerular inflammation. Because of the rash, I considered Henoch–Schönlein purpura and obtained a skin-biopsy specimen, which showed no IgA deposition. When the renal biopsy suggested lupus nephritis, I started the patient on steroid treatment; he had initial improvement, but his renal function continued to deteriorate, and I sent him to Dr. Kay..")
20
DR. JONATHAN KAY'S DIAGNOSIS This patient's urinary sediment contained red cells, suggesting glomerular disease. The results of a renal biopsy reportedly showed endocapillary proliferative glomerulonephritis with immune complexes containing IgG, IgM, and C3. The pathologist who interpreted the renal biopsy raised the possibility of either postinfectious glomerulonephritis or lupus nephritis.

21
Antistreptolysin O antibodies were not detected, which ruled out a diagnosis of poststreptococcal glomerulonephritis. Although the initial test for antinuclear antibodies was weakly positive, the test repeated at this hospital was negative. About 5 percent of patients with systemic lupus erythematosus present without detectable serum antinuclear antibodies; however, these patients typically do not have renal involvement.

22
The disease in the patient under discussion clinically involved arterioles, capillaries, and venules, since there was cutaneous leukocytoclastic vasculitis and glomerulonephritis. There were no pulmonary involvement to suggest Goodpasture's syndrome. Microscopic polyangiitis, Wegener's granulomatosis, and the Churg–Strauss syndrome are unlikely diagnoses in the absence of antineutrophil cytoplasmic antibodies. Thus, the most likely diagnosis was Henoch–Schönlein purpura or cryoglobulinemic vasculitis. IgA deposition in the skin, which is pathognomonic of Henoch– Schöelein purpura, was not present in this case.

23
Laboratory findings in this patient that support a diagnosis of cryoglobulinemic vasculitis include: Hypocomplementemia with low levels of C4 and the presence of rheumatoid factor without antinuclear antibodies.

24
Blood was drawn under appropriate conditions to assess for cryoglobulins. The cryocrit was 3 percent, with a cryoglobulin that consisted of homogeneous IgM kappa and heterogeneous IgG protein — Findings that established the diagnosis of type II mixed cryoglobulinemia.

25
The optimal way to detect cryoglobulines

26
20 ml blood, after 10h fast If you don’t take it in 37 degrees, cryo will mix with RBCs, and will disappear. In 37 degrees, RBC cloth and the cryo remain in the tube. Gradually it will precipitate.

28
The specimen must be maintained between 37°C and 41°C from the time of its withdrawal until the serum is isolated in the laboratory. After centrifugation at 37°C to remove red cells and fibrin, the serum is transferred at room temperature to a cryocrit tube, which then is stored at 4°C for a week and observed daily for seven days. If the serum becomes cloudy, the cryocrit tube is centrifuged in the cold and the cryocrit is measured. The cryoglobulin is washed and then assessed for monoclonality by immunofixation and for the presence of rheumatoid factor.

29
Type 1- monclonal IgM Type 2 – with RF activity (mixed)* monoclonal IgM and polyclonal IgG** Type 3- with RF activity (mixed) polyclonal IgM and IgG *RF- AB against the Fc of IgG ** Polyclonal antibodies are secreted by different B cells lineages. They react against a specific antigen, each identifying a different epitope.

31
Type 1 Single monoclonal gammopathy Underlying malignancy High cryo level May be asymptomatic of have hyperviscosity syndrome

32
Hyperviscosity syndrome Raynaude Arterial thrombosis Hemorrhage Types of hyperviscosity syndromes vary by pathology. Serum hyperviscosity may cause neurologic or ocular disorders Polycythemic hyperviscosity, results in reduced blood flow and increased organ congestion

33
Type 2 Associated usually with HCV (80%) but also seen in malignancy and autoimmune diseases. SPEP may not detect tne monoclonal component. Cryocrit >5%. Often asymptomatic(>50%) but may be associated with cryo immune complex vasculitis more than with hyperviscosity syndrome.
 but may be associated with cryo immune complex vasculitis more than with hyperviscosity syndrome..")
34
Skin vasculitis Glomerulonephritis Arthropathy Neuropathy Systemic necrotising vasculitis (e.g. gut vasculitis).
..")
35
Type 3 Less commonly associated with vasculitis than type 2. Usually associated with HCV. Cryocrit <5%. RF positivity may be only initial clue. Often asymptomatic but may be associated with cryo immune complex vasculitis more than with hyperviscosity syndrome.

36
Clinical manifestaions Cutaneous: Purpura Distal necrosis Urticaria Livedo Leg ulcers

38
Clinical manifestations Raynaud’s Acrocyanosis Joints Renal Neurologic Hemmorhages Arterial thrombosis

39
Gorevic et al, 1980 Lab values in Mixed RF-100% Low C3 or C4 -60% Normal SPEP-30% Type 2-32% Type 3-67%

40
Presenting manifestation of Mixed Purpura – 92%/100% (presentation/over all) Arthralgia Hepatic disease-55%/70% Renal disease, hypertension, edema Raynaud’s- 25%/25% Leg ulcers-22%/30% Sjogren syndrome -15%/15% Foot drop-7.5%/7.5% Paresthesias-5%-12% Thyroiditis -5%/5%
 Arthralgia Hepatic disease-55%/70% Renal disease, hypertension, edema Raynaud’s- 25%/25% Leg ulcers-22%/30% Sjogren syndrome -15%/15% Foot drop-7.5%/7.5% Paresthesias-5%-12% Thyroiditis -5%/5%")
42
Glomerulonephritis In all types Follows oncet of purpura, sometimes by years. Greatest renal injury is associated with type 2.-monoclonal IgM-RF depositions with IgG and C3.

43
Clinical manifestations of mixed in 170 patients ferri et al 2002 Age-51 (mean) F:M 2.8 Purpura-91% Arthralgia -83% Raynaud’s-34% Peripheral neuropathy-36% Renal disease- 31% Liver disease-70% Anti HCV-90%, Type2/3=2:1
 F:M 2.8 Purpura-91% Arthralgia -83% Raynaud’s-34% Peripheral neuropathy-36% Renal disease- 31% Liver disease-70% Anti HCV-90%, Type2/3=2:1")
44
Mixed cryo in HCV Prevalence 40-56% Most patients asymptomatic or arthralgia/fatigue Mixed cryo vasculitis 1-10%. Skin- PP, ulcers, digital necrosis Nerve- stocking gloves sensory axonal, MNM Kidney- MPRN, usually with other manifestations of vasculitis Other- Arthralgia, sjogren, Ly, PAN like vasculitis, ischemic abdominal disease

45
טיפול?

46
Treatment of HCV infection +vasculitis Cessation of antigenic stimulation: clearance of virus- IFN/RBV. Particularly in mild to moderate disease without organ threatening vasculitis. Avoid in sever symptomatic vasculitis as may exacerbate vasculitic manifestation. Suppress expression or remove cryo IS-CS+- Cytotoxic in severe organ threatening disease Plasmapheresis Anti CD 20, r/u HBV

47
Recommendations for the management of HCV associated MC טיפול אנטי וירלי PEG IFN/RBV לשקול ב HCV related MC Rituximab– לשקול בוסקוליטיס קשה, כיבים בעור, נוירופטיה פריפרית או GN. סטרואידים במינון גבוה במצבים קשים. פלסמפרזיס במצבים מסכני חיים או קשים ורפרקטורים. ציקלופוספמיד במצבים קשים ורפרקטורים עם פלסמפרזיס. Autoimmunity reviews 2011

48
Approach to treating cryoglobulinemic vasculitis E. william, 2012 Anti Viral therapy with PEG-IFN +RBV (if no CI) This approach results in sustaind virologic response in 40-50% of patients in genotype 1 and >80% in genotype 2,3. Anti viral therapy my suffice for the treatment of mild to moderate disease combined with low dose prednisone for signs and symptoms of vasculitis. Combination of RTX and anti viral agents may become the best choice for severe disease. RTX may be useful as first lile for organ/life threatening disease as an alternative to standart IS therapy(AZA, CYC).
 This approach results in sustaind virologic response in 40-50% of patients in genotype 1 and >80% in genotype 2,3. Anti viral therapy my suffice for the treatment of mild to moderate disease combined with low dose prednisone for signs and symptoms of vasculitis. Combination of RTX and anti viral agents may become the best choice for severe disease. RTX may be useful as first lile for organ/life threatening disease as an alternative to standart IS therapy(AZA, CYC)..")
49
Cryoglobulinemia אימונוגלובולין אחד או יותר בסרום שעובר פרסיפיטציה מתחת ל 37 מעלות ומתמוסס בחימום. סוג 1- מונוקלונל Ig לרוב IgG סוג 2- IgG + IgM monoclonal RF סוג 3- IgG + IgM polyclonal RF גיל עמידה, F:M 1:3 HCV חיובי ב 90% לויקוציטוקלסטיק וסקוליטיס בכלי דם קטנים. קליניקה- פורפורה כמעט בכל החולים, בפרזנטציה לרוב. חולשה, ארתרלגיה, מעורבות כבד, רנו, מונונויריטיס מולטיפלקס. כליה מעורבת בשליש, ממברנופרוליפרטיב

Similar presentations





>")








 Dr. Raid Jastania. Vasculitis Inflammation of the walls of the vessels Causes of inflammation: –Infectious, physical, chemical,>")







 is the sudden onset of: – Haematuria (macroscopic/microscopic)>")