Download presentation
Presentation is loading. Please wait.

1
Introduction to Systems Biology: Systems Medicine Charu G. Kumar, Ph.D. Research Assistant Professor Department of Bioengineering

2
At the end of this talk, you will be able to: Understand some aspects of systems biology. (15 min) Do statistical analyses for high-throughput data (20 min) –Gene set enrichment analysis Infer Gene-networks (50-60 min) –Inference methods –Tools for developing and visualizing gene networks Understand basic biochemical reaction modeling (20 min)
 Do statistical analyses for high-throughput data (20 min) –Gene set enrichment analysis Infer Gene-networks (50-60 min) –Inference methods –Tools for developing and visualizing gene networks Understand basic biochemical reaction modeling (20 min).")
3
Leaders in the field Karl Ludwig von Bertalanffy (1950) laid out the General Systems Theory Denis Noble Mihajlo Mesarovic (1968) formally inducted the term, ‘Systems Biology’. Jacob and Monod (1970) described the Feedback regulatory mechanism on a molecular level.
 described the Feedback regulatory mechanism on a molecular level..")
4
What is Systems Biology? Study of interactions between the components, and emergence of functions and behavior of that system. This behavior is emergent (more than the sum of its processes) and not reductionist (behavior completely defined as sum or difference of component interactions). It is the science of modeling and discovering the broader dynamic and complex relationships between molecules in cell types or model organisms.
 and not reductionist (behavior completely defined as sum or difference of component interactions). It is the science of modeling and discovering the broader dynamic and complex relationships between molecules in cell types or model organisms..")
5
Systems biology captures the dynamic nature of the biological processes by focusing on the interactions and their reconstruction Metabolites turnover within a minute Figure from Vogel, 2011; Schwanhausser et al. 2011 Time-scales: Metabolic networks (few seconds)<<protein networks (secs-mins) < GRNs (mins to hours)
<<protein networks (secs-mins) < GRNs (mins to hours).")
6
Top-down Reconstructions (e.g. GRNs) from high-throughput molecular ‘omics’ data (microarray, proteomics, rna-seq) using Inference Methods (non-stoichiometric and coarse-grained)
 from high-throughput molecular ‘omics’ data (microarray, proteomics, rna-seq) using Inference Methods (non-stoichiometric and coarse-grained).")
7
Protein and GR networks Figures taken from Sauro’s book on Control theory for biologists Protein mechanisms Gene regulatory patterns

8
Bottom-up Reconstructions using direct methods for generating metabolic models (stoichiometric networks)
")
9
Tailoring to tissues; Drug response phenotypes Adaptive evolution; Disease progression; Synthetic biology; Metabolic engineering Systems Biology Paradigm: components networks computational models phenotypes Figure adpated from Nathan Price’s course slides

10
Processing of high-throughput ‘omics’ data Noise filtering, background correction (adjust data for background intensity surrounding each feature i.e. non-specific hybridization) Normalization (adjusting values for spatial heterogeneity, different dye absorption, etc in samples) Feature summarization. Can use GenePattern for microarray analysis : http://www.broadinstitute.org/cancer/software/genepattern/
 Normalization (adjusting values for spatial heterogeneity, different dye absorption, etc in samples) Feature summarization. Can use GenePattern for microarray analysis :")
11
Gene Set Enrichment analysis (GSEA) HT anal results in gene lists that we evaluate using our favorite statistical test (Hypergeometric, t-test, Z- test etc) which give a p-value = P(this sample |Ho is true). For multiple comparisons, p-value adjusted for false discovery (q-value). Alternate tool developed by Subramanian et al (2005) Is your gene list over-represented in some known gene set (published gene list representing a pathway or GO category, or cytogenetic bands)? Needs these files: –Entire microarray data (Specific defined formats) –Sample phenotype –Known gene set –Microarray Chip annotation
. Alternate tool developed by Subramanian et al (2005) Is your gene list over-represented in some known gene set (published gene list representing a pathway or GO category, or cytogenetic bands). Needs these files: –Entire microarray data (Specific defined formats) –Sample phenotype –Known gene set –Microarray Chip annotation.")
12
GSEA input files (*.GCT)
")
13
GSEA input: Phenotype.cls file

14
GenePattern can convert RNA-seq (and other format files) into.gct
 into.gct")
15
GSEA params

16
GO category Gene clusters ES(S) is calculated based on both the correlations and the positions in ranked matrix. Genes in expression matrix are sorted based on correlation to phenotype classes Compute ES for each permutation. Mootha et al. Nature Genetics, 2003 Compare the distribution of these ES with ES for actual data.

18
GSEA: Leading Edge Analysis http://www.broadinstitute.org/gsea/doc/GSEAUserGuideFrame.html

19
Top-down Reconstructions (e.g. GRNs) from high-throughput molecular ‘omics’ data (microarray, proteomics, rna-seq) using Inference Methods GSEA
 from high-throughput molecular ‘omics’ data (microarray, proteomics, rna-seq) using Inference Methods GSEA.")
20
Networks are mathematical graphs consisting of nodes, and edges joining those nodes. The degree or connectivity ‘d’ of a node is the number of edges from that node. Power-law distribution; P(d) ∝ d -γ, γ is a constant that is characteristic of the network
 ∝ d -γ, γ is a constant that is characteristic of the network.")
21
Gene Regulatory Networks Describe the interaction between TFs (and/or miRNA) and genes. GRNs are information processing networks that help determine the rate of protein production. Inouye and Kaneko, PLoS Comp. Biol. 2013 Xj->Y X Y Rate of production Y =f (X*)
.")
22
Kumar et al. 2010 BMC Genomics 11:161 Agglomerative hierarchical clustering with average correlation >0.75 Used Match to search TRANFAC db; Each TFBS in cluster tested for significant enrichment. ANN-Spec for motif prediction; Tomtom

23
CARMAweb (https://carmaweb.genome.tugraz.at/carma/)

24
Unsupervised methods for inference of GRNs The Algorithm for the Reconstruction of Accurate Cellular Networks(ARACNE) –Margolin et al. 2006. BMC Bioinformatics 7: S7 Context Likelihood Relatedness (CLR) –Faith et al. 2007. PLOS Biol. The above two methods are based on Mutual Information (MI) for identifying co-expression networks. MI measures the dependency between two random variables i.e. to what extent does one variable reduce the uncertainty of prediction in the other. Weighted Gene Co-expression Network Analysis (WGCNA) WGCNA is based on Pearson correlation
 –Faith et al PLOS Biol. The above two methods are based on Mutual Information (MI) for identifying co-expression networks. MI measures the dependency between two random variables i.e. to what extent does one variable reduce the uncertainty of prediction in the other. Weighted Gene Co-expression Network Analysis (WGCNA) WGCNA is based on Pearson correlation.")
25
ARACNE Works with more than 100 microarray samples Basso et al. 2005 DPI: I(g 1, g 3 ) ≤ min [I(g 1, g 2 ); I(g 2, g 3 )] Finds the weakest link of a triplet Removes that edge. Infers the most likely path of information flow.
 ≤ min [I(g 1, g 2 ); I(g 2, g 3 )] Finds the weakest link of a triplet Removes that edge. Infers the most likely path of information flow..")
26
ARACNE download and setup aracne –i /data/input.exp –k 0.15 –t 0.05–r 1 http://wiki.c2b2.columbia.edu/califanolab/index.php/Software/ARACNE aracne2 –i /data/input.exp –k 0.15 –t 0.05 –r 1 –p 1e-7 Outputs an adjacency matrix that consists of inferred interactions. To view the adjacency matrix as a network, geWorkbench can be installed from https://gforge.nci.nih.gov/frs/?group_id=78https://gforge.nci.nih.gov/frs/?group_id=78 Nature protocol has tutorial, manual and technical report Margolin et al. Nature Protocols 1, - 662 - 671 (2006)
.")
27
Aracne command line

28
JAVA GUI geWorkbench http://wiki.c2b2.columbia.edu/workbench/index.php/Project_Folders

29
Network visualization (Cytoscape component) geWorkbench can be installed from https://gforge.nci.nih.gov/frs/?group_id=78 https://gforge.nci.nih.gov/frs/?group_id=78
 geWorkbench can be installed from group_id=78 group_id=78")
30
Simple Interaction format (SIF) for Cytoscape nodeA nodeB nodeC nodeA nodeD nodeE... nodeY nodeZ
 for Cytoscape nodeA nodeB nodeC nodeA nodeD nodeE... nodeY nodeZ")
31
Metabolic models are stoichiometric representations of all possible biochemical reactions in the cell. 1.Provide a mapping between genotype and the phenotype 2.Identify key features of metabolism such as growth yield, network robustness, and gene essentiality. 3.Models of yeast have been used to investigate production of therapeutic proteins, as yeast model allow modeling of PTMs. 4.Pathogenic models allow for development of novel drugs to combat infection with minimal side-effects to host. 5.Metabolic models of mammals have been employed to study various diseases. 6.Model microbes for their biotechnological applications, such as fermentation, biofuel production, etc.

32
Kim TY et al. 2012

33
PATHOLOGIC, the model SEED Figure adapted from Nathan Price’s course slides

34
Step 2: Refinement of reconstruction Verify rxns for enzyme and substrate specificity, Gene-Protein- Reaction formulation, stoichiometry, directionality, and location. Figure adapted from Nathan Price’s course slides

35
Figure from Nathan Price’s course slides

36
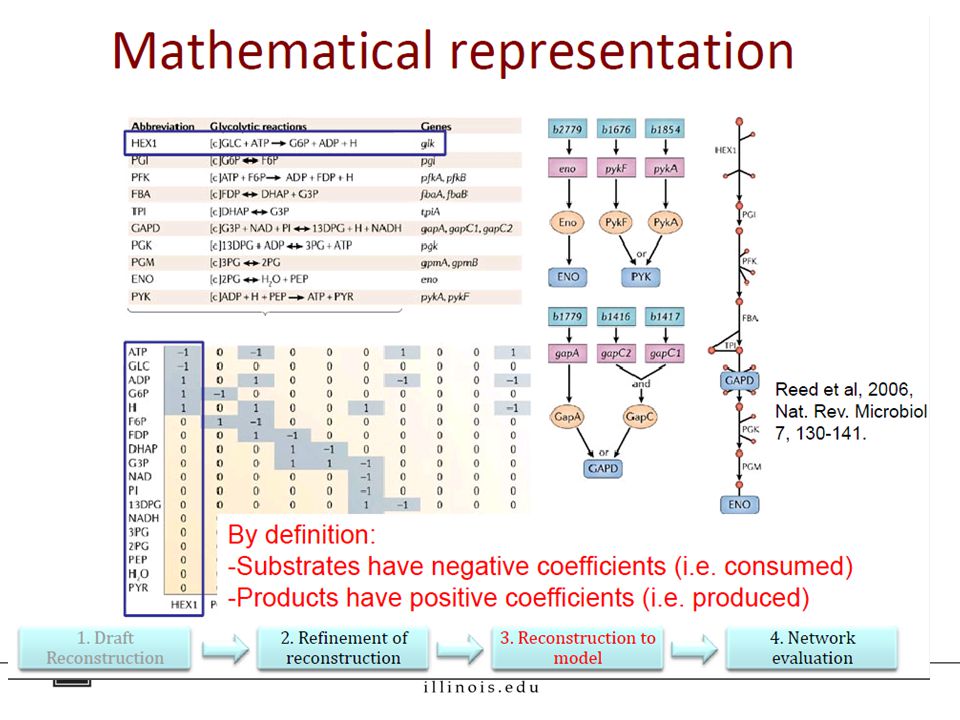
Step 3: Converting the reconstruction into a computable form. Mathematically represent the reconstruction as a matrix Define system boundaries [extracellular, intracellular, and exchange reactions e.g. transport, which are represented w.r.t the extracellular environment (secretion is +ve flux, uptake is –ve flux)]. Add constraints Mass balance Steady ‐ state Thermodynamics (e.g., reaction directionality) Environmental constraints (e.g. presence or absence of nutrients) Regulatory (e.g., on/off gene expression )
]. Add constraints Mass balance Steady ‐ state Thermodynamics (e.g., reaction directionality) Environmental constraints (e.g. presence or absence of nutrients) Regulatory (e.g., on/off gene expression ).")
37
= S matrix

39
Metabolic model consists of three components The reaction network, which is encoded as a stoichiometric matrix [parsed using the COBRA toolbox].stoichiometric matrix A list of rules called gene-protein-reaction (GPR) associations that describe how gene activity is linked to reaction activity. A biomass function, which is a list of small molecules, co- factors, nucleotides, amino acids, lipids, and cell wall components needed to support growth and division. Assumption used for modeling: Metabolism is in steady state. i.e. Uptake and secretion have reached a plateau; d[A]/dt ≈ 0
![Metabolic model consists of three components The reaction network, which is encoded as a stoichiometric matrix [parsed using the COBRA toolbox].stoichiometric matrix A list of rules called gene-protein-reaction (GPR) associations that describe how gene activity is linked to reaction activity.](http://images.slideplayer.com/14/4236967/slides/slide_39.jpg "A biomass function, which is a list of small molecules, co- factors, nucleotides, amino acids, lipids, and cell wall components needed to support growth and division. Assumption used for modeling: Metabolism is in steady state. i.e. Uptake and secretion have reached a plateau; d[A]/dt ≈ 0.")
40
Mathematically, the S matrix is a linear transformation of the unsolved flux vector v = (v 1,v 2,.., v n ) to a vector of time derivatives of the concentration vector x = (x 1, x 2,.., x m ) as = S ∙ v V 1 V 2 A -1 0 Rxns for B: A ↔B, V 1 ; 2B ↔ C, V 2 ; B 1 -2 Mass balance: ; C 0 1 Steady state: At steady state, the change in concentration as a function of time is zero; hence, dx/dt = S ∙ v = 0 Solve for the possible set of flux vectors. Flux Balance Analysis

41
Constraints and Biomass Objective Function The set of possible flux vectors are further constrained by defining v i(lb) ≤ v i ≤ v i(ub) for reaction i. Assume Objective of organisms: grow, divide and proliferate. Need biomass generating metabolic precursors (e.g. aa, nts, phospholipids, vit., cofactors, energy req). This Biomass Objective Function requires dry cell weight composition, and macromolecular breakdown. For 1gDW Ecoli Adapted from Nogales et al. BMC Sys Biol. 2008 Z = 41.257v ATP - 3.547v NADH + 18.225v NADPH + 0.205v G6P + 0.0709v F6P +0.8977v R5P + 0.361v E4P + 0.129v T3P + 1.496v 3PG + 0.5191v PEP +2.8328v PYR + 3.7478v AcCoA + 1.7867v OAA + 1.0789v AKG Using steady state fluxes, solve using linear programming to optimize Z.
. This Biomass Objective Function requires dry cell weight composition, and macromolecular breakdown. For 1gDW Ecoli Adapted from Nogales et al. BMC Sys Biol Z = v ATP v NADH v NADPH v G6P v F6P v R5P v E4P v T3P v 3PG v PEP v PYR v AcCoA v OAA v AKG Using steady state fluxes, solve using linear programming to optimize Z..")
42
Figure adapted from Nathan Price’s course slides

43
Step 4: Evaluation of network content Evaluate content pathway by pathway Will ease identification of missing genes & reactions Draw metabolic maps to ease detection of missing rxns –Gap analysis e.g. H.pylori has 2 of 4 enzymes missing for Ile and Val synthesis. Gap? No. Turns out Ile, Val are needed in medium to grow. Analysis of dead-end metabolites (either consumed OR produced) Network evaluation: can it generate biomass components, precursors to metabolites, mass-charge balancing, etc.
 Network evaluation: can it generate biomass components, precursors to metabolites, mass-charge balancing, etc..")
44
Conclusions Top down reconstruction: of networks using high- throughput data requires reliable statistical predictions. Gene Set Enrichment Analysis is an alternative to looking for over-representation in your gene list, by looking for enrichment of genes in defined gene sets. Gene regulatory network inference using Aracne Bottom up reconstructions: result in a more precise, mathematically d(r)efined model.
efined model..")
45
References 1: Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005 Oct 25;102(43):15545-50. Epub 2005 Sep 30. PubMed PMID: 16199517; PubMed Central PMCID: PMC1239896. 2: Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003 Jul;34(3):267-73. PubMed PMID: 12808457. 3: Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, Cottarel G, Kasif S, Collins JJ, Gardner TS. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007 Jan;5(1):e8. PubMed PMID: 17214507; PubMed Central PMCID: PMC1764438. 4: Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A. Reverse engineering of regulatory networks in human B cells. Nat Genet. 2005 Apr;37(4):382-90. Epub 2005 Mar 20. PubMed PMID: 15778709. 5: Kumar CG, Everts RE, Loor JJ, Lewin HA. Functional annotation of novel lineage-specific genes using co-expression and promoter analysis. BMC Genomics. 2010 Mar 9;11:161. doi: 10.1186/1471- 2164-11-161. PubMed PMID: 20214810; PubMed Central PMCID: PMC2848242. 6: Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011 May 19;473(7347):337-42. doi: 10.1038/nature10098. Erratum in: Nature. 2013 Mar 7;495(7439):126-7. PubMed PMID: 21593866. Thiele I, Palsson BØ. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 2010 Jan;5(1):93-121. doi:10.1038/nprot.2009.203. Epub 2010 Jan 7. PubMed PMID: 20057383; PubMed Central PMCID: PMC3125167.
: Epub 2005 Sep 30. PubMed PMID: ; PubMed Central PMCID: PMC : Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet Jul;34(3): PubMed PMID: : Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, Cottarel G, Kasif S, Collins JJ, Gardner TS. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol Jan;5(1):e8. PubMed PMID: ; PubMed Central PMCID: PMC : Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A. Reverse engineering of regulatory networks in human B cells. Nat Genet Apr;37(4): Epub 2005 Mar 20. PubMed PMID: : Kumar CG, Everts RE, Loor JJ, Lewin HA. Functional annotation of novel lineage-specific genes using co-expression and promoter analysis. BMC Genomics Mar 9;11:161. doi: / PubMed PMID: ; PubMed Central PMCID: PMC : Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature May 19;473(7347): doi: /nature Erratum in: Nature Mar 7;495(7439): PubMed PMID: Thiele I, Palsson BØ. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc Jan;5(1): doi: /nprot Epub 2010 Jan 7. PubMed PMID: ; PubMed Central PMCID: PMC")
46
Thank you!

47
Tools for Enrichment analysis DAVID BinGO (Cytoscape app) GSEA GoMiner: http://discover.nci.nih.gov/gominer GOstat: http://gostat.wehi.edu.au
 GSEA GoMiner: GOstat:")
Similar presentations



 Presented By: Dan Baluta.>")

>")



 Speack: Zhu YANG 2006.10.04.>")

 Fitness.>")






 DNA Microarray, 2d gel, MSMS, yeast 2-hybrid.>")
 Analysis of co-expression Search for similarly expressed genes experiment1 experiment2 experiment3 ……….. Gene i:>")
