Download presentation
Presentation is loading. Please wait.

1
Nanocomposites: mixing CNTs into polymers

3
Outline 1.Introduction 2. Composites of multiwalled carbon nanotubes (MWNT) with polycarbonate (PC) produced by masterbatch dilution technique Electrical resistivity Dispersion and alignment Influence of processing parameters on electrical resistivity 3. Composites of MWNT and SWNT with PC produced by direct incorporation Percolation of different commercial MWNT in PC Percolation of SWNT in PC Stress-strain behaviour 4. Summary
 with polycarbonate (PC) produced by masterbatch dilution technique Electrical resistivity Dispersion and alignment Influence of processing parameters on electrical resistivity 3. Composites of MWNT and SWNT with PC produced by direct incorporation Percolation of different commercial MWNT in PC Percolation of SWNT in PC Stress-strain behaviour 4. Summary.")
4
–Electrical conductivity –Improvement of mechanical properties, especially strength –Enhancement of thermal stability –Enhancement of thermal conductivity –Improvement of fire retardancy –Enhancement of oxidation stability –Effects at low CNT contents because of the very high aspect ratio Benefits of CNTs to polymers

5
How to introduce CNTs into polymers

6
Melt mixing of CNT with thermoplastic polymers

7
Preparation of the PC-MWNT composites Masterbatch technology: polycarbonate(PC) + PC based masterbatch (15 wt% MWNT) –masterbatch (Hyperion Catalysis International, Inc, Cambridge, USA) diluted with PC Iupilon E2000 (PC1), PC Lexan 121 (PC2) or PC as used for the masterbatch (PC3) –Haakeco-rotating, intermeshing twin screw extruder with one kilogramm mixtures –DACA Micro Compounder, conical twin screw extruder (4.5 cm 3 capacity) –Brabender PL-19 single screw extruder
 + PC based masterbatch (15 wt% MWNT) –masterbatch (Hyperion Catalysis International, Inc, Cambridge, USA) diluted with PC Iupilon E2000 (PC1), PC Lexan 121 (PC2) or PC as used for the masterbatch (PC3) –Haakeco-rotating, intermeshing twin screw extruder with one kilogramm mixtures –DACA Micro Compounder, conical twin screw extruder (4.5 cm 3 capacity) –Brabender PL-19 single screw extruder")
8
Characterization of the masterbatch (PC + 15 wt% MWNT)
")
9
Dispersion in PC-MWNT composites

10
Alignment in PC-MWNT composites

11
Comparison for different set of PC masterbatch dilution

12
Detection of percolation and influence of processing conditions investigated by dielectric spectroscopy

13
Direct incorporation of different kinds of commercial MWNT into PC

14
Comparison of direct incorporation of CNT, masterbatch dilution, and CB addition

15
Direct incorporationof SWNT1 into PC

18
Direct incorporationof SWNT2 into PC

20
Summary Melt mixing is a powerful method to disperse CNT into polymers Masterbatch dilution technique (based on a PC masterbatch) –percolation in the range of 1.0 wt% MWNT –suitable processing conditions can shift percolation to lower values (0.5wt%) –effects of mixing equipment and PC viscosity on percolation are small Direct incorporation method –percolation strongly depends on the kind of CNT, production method (resulting in different sizes, purity and defect levels), and the purifying/modification steps –for commercial MWNT percolation occurs between 1.0 and 3.0 wt% and is lower at lower MWNT diameters and higher purity –HipCO-SWNT (CNI) percolation between 0.30 and 0.35 wt% –stress-strain behavior of the composites: modulus and stress are enhanced, elongation at break reduced especially above percolation concentration
 –percolation in the range of 1.0 wt% MWNT –suitable processing conditions can shift percolation to lower values (0.5wt%) –effects of mixing equipment and PC viscosity on percolation are small Direct incorporation method –percolation strongly depends on the kind of CNT, production method (resulting in different sizes, purity and defect levels), and the purifying/modification steps –for commercial MWNT percolation occurs between 1.0 and 3.0 wt% and is lower at lower MWNT diameters and higher purity –HipCO-SWNT (CNI) percolation between 0.30 and 0.35 wt% –stress-strain behavior of the composites: modulus and stress are enhanced, elongation at break reduced especially above percolation concentration")
21
Graphene–polymer composite Graphite oxide was prepared by the Hummers method from SP-1 graphite (Bay Carbon), and dried for a week over phosphorus pentoxide in a vacuum desiccator. Dried graphite oxide (50 mg) was suspended in anhydrous DMF (5 ml, Dow-Grubbs solvent system), treated with phenyl isocyanate (2 mmol, Sigma-Aldrich) for 24 h, and recovered by filtration through a sintered glass funnel (50 ml, medium porosity). Stable dispersions of the resulting phenyl isocyanate-treated graphite oxide materials were prepared by ultrasonic exfoliation (Fisher Scientific FS60, 150 W, 1 h) in DMF (1 mg ml-1). Polystyrene (Scientific Polymer Products, approximate Mw = 280 kD, PDI = 3.0) was added to these dispersions and dissolved with stirring (Fig. 1d, left). Reduction of the dispersed material (Fig. 1d, right) was carried out with dimethylhydrazine (0.1 ml in 10 ml of DMF, Sigma-Aldrich) at 80 °C for 24 h. Upon completion, the coagulation of the polymer composites was accomplished by adding the DMF solutions dropwise into a large volume of vigorously stirred methanol (10:1 with respect to the volume of DMF used). The coagulated composite powder (Fig. 1e) was isolated via filtration; washed with methanol (200 ml); dried at 130 °C under vacuum for 10 h to remove residual solvent, anti-solvent, and moisture; crushed into a fine powder with a mortar and pestle, and then pressed (Fig. 1f) in a hydraulic hot press (Model 0230C-X1, PHI-Tulip) at 18 kN with a temperature of 210 °C.Fig. 1d Fig. 1eFig. 1f
 was suspended in anhydrous DMF (5 ml, Dow-Grubbs solvent system), treated with phenyl isocyanate (2 mmol, Sigma-Aldrich) for 24 h, and recovered by filtration through a sintered glass funnel (50 ml, medium porosity). Stable dispersions of the resulting phenyl isocyanate-treated graphite oxide materials were prepared by ultrasonic exfoliation (Fisher Scientific FS60, 150 W, 1 h) in DMF (1 mg ml-1). Polystyrene (Scientific Polymer Products, approximate Mw = 280 kD, PDI = 3.0) was added to these dispersions and dissolved with stirring (Fig. 1d, left). Reduction of the dispersed material (Fig. 1d, right) was carried out with dimethylhydrazine (0.1 ml in 10 ml of DMF, Sigma-Aldrich) at 80 °C for 24 h. Upon completion, the coagulation of the polymer composites was accomplished by adding the DMF solutions dropwise into a large volume of vigorously stirred methanol (10:1 with respect to the volume of DMF used). The coagulated composite powder (Fig. 1e) was isolated via filtration; washed with methanol (200 ml); dried at 130 °C under vacuum for 10 h to remove residual solvent, anti-solvent, and moisture; crushed into a fine powder with a mortar and pestle, and then pressed (Fig. 1f) in a hydraulic hot press (Model 0230C-X1, PHI-Tulip) at 18 kN with a temperature of 210 °C.Fig. 1d Fig. 1eFig. 1f.")
23
Process flow of graphene– polymer composite fabrication a, SEM and digital image (inset) of natural graphite. b, A typical AFM non-contact-mode image of graphite oxide sheets deposited onto a mica substrate from an aqueous dispersion (inset) with superimposed cross-section measurements taken along the red line indicating a sheet thickness of 1 nm. c, AFM image of phenyl isocyanate-treated graphite oxide sheets on mica and profile plot showing the 1 nm thickness. d, Suspension of phenyl isocyanate- treated graphite oxide (1 mg ml-1) and dissolved polystyrene in DMF before (left) and after (right) reduction by N,N-dimethylhydrazine. e, Composite powder as obtained after coagulation in methanol. f, Hot- pressed composite (0.12 vol.% of graphene) and pure polystyrene of the same 0.4-mm thickness and processed in the same way. g, Low (top row) and high (bottom row) magnification SEM images obtained from a fracture surface of composite samples of 0.48 vol.% (left) and 2.4 vol.% (right) graphene in polystyrene.
 with superimposed cross-section measurements taken along the red line indicating a sheet thickness of 1 nm. c, AFM image of phenyl isocyanate-treated graphite oxide sheets on mica and profile plot showing the 1 nm thickness. d, Suspension of phenyl isocyanate- treated graphite oxide (1 mg ml-1) and dissolved polystyrene in DMF before (left) and after (right) reduction by N,N-dimethylhydrazine. e, Composite powder as obtained after coagulation in methanol. f, Hot- pressed composite (0.12 vol.% of graphene) and pure polystyrene of the same 0.4-mm thickness and processed in the same way. g, Low (top row) and high (bottom row) magnification SEM images obtained from a fracture surface of composite samples of 0.48 vol.% (left) and 2.4 vol.% (right) graphene in polystyrene..")
24
Advantages of Nanosized Additions The Nanocomposites 2000 conference has revealed clearly the property advantages that nanomaterial additives can provide in comparison to both their conventional filler counterparts and base polymer. Properties which have been shown to undergo substantial improvements include: Mechanical properties e.g. strength, modulus and dimensional stability Decreased permeability to gases, water and hydrocarbons Thermal stability and heat distortion temperature Flame retardancy and reduced smoke emissions Chemical resistance Surface appearance Electrical conductivity Optical clarity in comparison to conventionally filled polymers

25
Disadvantages of Nanosized Additions To date one of the few disadvantages associated with nanoparticle incorporation has concerned toughness and impact performance. Some of the data presented has suggested that nanoclay modification of polymers such as polyamides, could reduce impact performance. Clearly this is an issue which would require consideration for applications where impact loading events are likely. In addition, further research will be necessary to, for example, develop a better understanding of formulation/structure/property relationships, better routes to platelet exfoliation and dispersion etc.

26
Examples of Mechanical Property gains due to Nanoparticle Additions Data provided by Hartmut Fischer of TNO in the Netherlands relating to polyamide – montmorillonite nanocomposites indicates tensile strength improvements of approximately 40 and 20% at temperatures of 23ºC and 120ºC respectively and modulus improvements of 70% and a very impressive 220% at the same temperatures. In addition Heat Distortion Temperature was shown to increase from 65ºC for the unmodified polyamide to 152ºC for the nanoclay-modified material, all the above being achieved with just a 5% loading of montmorillonite clay. Similar mechanical property improvements were presented for polymethyl methacrylate – clay hybrids. Further data provided by Akkepeddi of Honeywell relating to polyamide-6 polymers confirms these property trends. In addition, the further benefits of short/long glass fibre incorporation, together with nanoclay incorporation, are clearly revealed.

27
Area of Applications Such mechanical property improvements have resulted in major interest in nanocomposite materials in numerous automotive and general/industrial applications. These include potential for utilization as mirror housings on various vehicle types, door handles, engine covers and intake manifolds and timing belt covers. More general applications currently being considered include usage as impellers and blades for vacuum cleaners, power tool housings, mower hoods and covers for portable electronic equipment such as mobile phones, pagers etc.

28
Gas Barrier The gaseous barrier property improvement that can result from incorporation of relatively small quantities of nanoclay materials is shown to be substantial. Data provided from various sources indicates oxygen transmission rates for polyamide-organoclay composites which are usually less than half that of the unmodified polymer. Further data reveals the extent to which both the amount of clay incorporated in the polymer, and the aspect ratio of the filler contributes to overall barrier performance. In particular, aspect ratio is shown to have a major effect, with high ratios (and hence tendencies towards filler incorporation at the nano-level) quite dramatically enhancing gaseous barrier properties. Such excellent barrier characteristics have resulted in considerable interest in nanoclay composites in food packaging applications, both flexible and rigid. Specific examples include packaging for processed meats, cheese, confectionery, cereals and boil-in-the-bag foods, also extrusion- coating applications in association with paperboard for fruit juice and dairy products, together with co-extrusion processes for the manufacture of beer and carbonated drinks bottles. The use of nanocomposite formulations would be expected to enhance considerably the shelf life of many types of food.
 quite dramatically enhancing gaseous barrier properties. Such excellent barrier characteristics have resulted in considerable interest in nanoclay composites in food packaging applications, both flexible and rigid. Specific examples include packaging for processed meats, cheese, confectionery, cereals and boil-in-the-bag foods, also extrusion- coating applications in association with paperboard for fruit juice and dairy products, together with co-extrusion processes for the manufacture of beer and carbonated drinks bottles. The use of nanocomposite formulations would be expected to enhance considerably the shelf life of many types of food..")
29
Fuel Tanks The ability of nanoclay incorporation to reduce solvent transmission through polymers such as polyamides has been demonstrated. Data provided by De Bievre and Nakamura of UBE Industries reveals significant reductions in fuel transmission through polyamide–6/66 polymers by incorporation of a nanoclay filler. As a result, considerable interest is now being shown in these materials as both fuel tank and fuel line components for cars. Of further interest for this type of application, the reduced fuel transmission characteristics are accompanied by significant material cost reductions.

30
Films The presence of filler incorporation at nano-levels has also been shown to have significant effects on the transparency and haze characteristics of films. In comparison to conventionally filled polymers, nanoclay incorporation has been shown to significantly enhance transparency and reduce haze. With polyamide based composites, this effect has been shown to be due to modifications in the crystallisation behaviour brought about by the nanoclay particles; spherilitic domain dimensions being considerably smaller. Similarly, nano-modified polymers have been shown, when employed to coat polymeric transparency materials, to enhance both toughness and hardness of these materials without interfering with light transmission characteristics. An ability to resist high velocity impact combined with substantially improved abrasion resistance was demonstrated by Haghighat of Triton Systems.

31
Environmental Protection Water laden atmospheres have long been regarded as one of the most damaging environments which polymeric materials can encounter. Thus an ability to minimize the extent to which water is absorbed can be a major advantage. Data provided by Beall from Missouri Baptist College indicates the significant extent to which nanoclay incorporation can reduce the extent of water absorption in a polymer. Similar effects have been observed by van Es of DSM with polyamide based nanocomposites. In addition, van Es noted a significant effect of nanoclay aspect ratio on water diffusion characteristics in a polyimide nanocomposite. Specifically, increasing aspect ratio was found to diminish substantially the amount of water absorbed, thus indicating the beneficial effects likely from nanoparticle incorporation in comparison to conventional microparticle loading. Hydrophobic enhancement would clearly promote both improved nanocomposite properties and diminish the extent to which water would be transmitted through to an underlying substrate. Thus, applications in which contact with water or moist environments is likely could clearly benefit from materials incorporating nanoclay particles.

32
Preparation and Characterization of Novel Polymer/Silicate Nanocomposites Five categories cover the majority of composites synthesized with more recent techniques being modifications or combinations from this list. Type I: Organic polymer embedded in an inorganic matrix without covalent bonding between the components. Type II: Organic polymer embedded in an inorganic matrix with sites of covalent bonding between the components.

33
Preparation and Characterization of Novel Polymer/Silicate Nanocomposites Type III: Co-formed interpenetrating networks of inorganic and organic polymers without covalent bonds between phases. Type IV: Co-formed interpenetrating networks of inorganic and organic polymers with covalent bonds between phases. Type V: Non-shrinking simultaneous polymerization of inorganic and organic polymers.

34
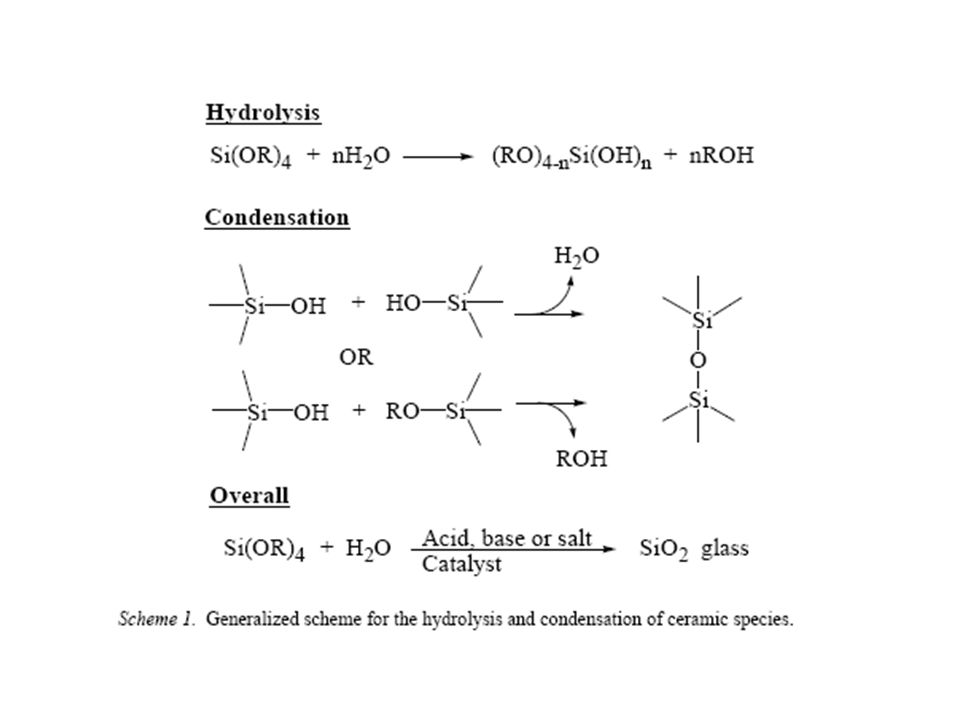
Preparation and Characterization of Novel Polymer/Silicate Nanocomposites The great majority of nanocomposites incorporate silica from tetraethoxysilane (TEOS). The formation of the inorganic component involves two steps, hydrolysis and condensation as seen in Scheme 1.

36
Polymers considered: PEO, PEO/PPO, PVAc, PVA, PAN, MEEP A general synthesis for a base, acid, or salt catalyzed polyphosphazene, polyethylene oxide (PEO), and polyethylene oxide/polypropylene oxide (PPO/PEO) block nanocomposite is as follows: 300 mg of polymer is dissolved into 10 mL of a 50/50 by volume tetrahydrofuran (THF)/ethanol mixed solvent in a capped vial. To this solution is added TEOS (336 mg). A catalyst is then introduced as an aqueous solution (150 μl) and the mixture is capped and sonicated at 50 o C for 30 minutes. The solution is aged from hours to days depending upon the catalyst used in a sealed vial and poured into a Teflon mold and loosely covered at room temperature. The nanocomposite self assembles as the volatile solvent slowly escapes during the condensation process. The synthesis of polyvinyl acetate (PVAc)/silicate nanocomposites requires a different approach from the other nanocomposites. PVAc (300 mg) is dissolved into an 50/50 by volume acetic acid/methanol (10 mL) mixed solvent in a capped vial. To this solution is added TEOS (373 mg). The solution is then sonicated for 5 minutes in a sealed vial at room temperature and poured into a Teflon mould and loosely covered at room temperature. The nanocomposite self assembles during the curing process, which typically lasts up to 24 hours. Additional heating at 100 °C for 30 minutes aids in removing lingering acetic acid from the nanocomposite.
. A catalyst is then introduced as an aqueous solution (150 μl) and the mixture is capped and sonicated at 50 o C for 30 minutes. The solution is aged from hours to days depending upon the catalyst used in a sealed vial and poured into a Teflon mold and loosely covered at room temperature. The nanocomposite self assembles as the volatile solvent slowly escapes during the condensation process. The synthesis of polyvinyl acetate (PVAc)/silicate nanocomposites requires a different approach from the other nanocomposites. PVAc (300 mg) is dissolved into an 50/50 by volume acetic acid/methanol (10 mL) mixed solvent in a capped vial. To this solution is added TEOS (373 mg). The solution is then sonicated for 5 minutes in a sealed vial at room temperature and poured into a Teflon mould and loosely covered at room temperature. The nanocomposite self assembles during the curing process, which typically lasts up to 24 hours. Additional heating at 100 °C for 30 minutes aids in removing lingering acetic acid from the nanocomposite..")
37
Applications One of the most interesting of these applications is as solid polymer electrolytes (SPE) for lithium batteries. The polyphosphazene MEEP is a well-known SPE with very high room temperature conductivity, however it lacks the mechanical stability to be used in a practical device (12). Traditional stabilization methods, such as deep UV or electron beam crosslinking methods do improve the physical stability of SPEs, however this crosslinking lowers ionic conductivity – tests performed in our laboratory revealed this to be a factor of 30-45 for MEEP-like phosphazene polymers. This reduction is due to the additional covalent linkages formed during the crosslinking process that inhibit chain segmental motion and ion transfer. Since the nanocomposites formed by the ceramic condensation process do not form bonds to the polymer component, (Type I nanocomposites) mechanical stabilization is achieved without a great loss of ionic conductivity (13). However, these nanocomposites have the highest tensile strength of any of the catalyst types studied; yet they were also found to be glassy and brittle.
. Traditional stabilization methods, such as deep UV or electron beam crosslinking methods do improve the physical stability of SPEs, however this crosslinking lowers ionic conductivity – tests performed in our laboratory revealed this to be a factor of for MEEP-like phosphazene polymers. This reduction is due to the additional covalent linkages formed during the crosslinking process that inhibit chain segmental motion and ion transfer. Since the nanocomposites formed by the ceramic condensation process do not form bonds to the polymer component, (Type I nanocomposites) mechanical stabilization is achieved without a great loss of ionic conductivity (13). However, these nanocomposites have the highest tensile strength of any of the catalyst types studied; yet they were also found to be glassy and brittle..")
38
Goal for Type I Nanocomposites The goal in the process is to form a completely interpenetrating network (IPN) of both inorganic and organic phases. Homogeneous nanocomposites with good IPNs are often stronger, more resilient, and optically transparent, whereas heterogeneous composites are often mechanically weaker and opaque.

39
Novel Rubber Nanocomposites with Adaptable Mechanical Properties Silica particles have become more important in tire applications since the introduction of the Green Tire® by Michelin. As a filler, silica has greater reinforcing power, such as improving tear strength, abrasion resistance, age resistance and adhesion properties than carbon black [6-8]. However, due to the strong inter-particle hydrogen bonds between hydroxyl groups, the agglomeration nature of silica is generally believed to be responsible for the significant Payne effect which brings about considerable rolling resistance for tire applications. In order to reduce the filler-filler interaction and/or to enhance the mechanical properties of silica filled composites, researchers have been working for many years on different strategies to improve silica-rubber interaction and, in turn, to reduce the rolling resistance. Among these strategies, chemical modifications of rubbers by attaching functional groups interacting with silica [9-22] and surface treatments of silica by reducing surface polarity with different silane coupling agents [22-36] are the most popular techniques.

40
Novel Rubber Nanocomposites with Adaptable Mechanical Properties However, these techniques admittedly have quite a few drawbacks. For the former technique, the chemical modification reaction of rubber was usually not applicable to commercial production and its degree of modification was usually very low [9,11,14,18,22]. Additionally, the chemical modification was limited to rubber chain ends [12,17,20], meaning that the final silica composite was unsatisfactory in terms of reducing silica agglomeration. For the latter, the used coupling agents are expensive and it could possibly lower the crosslinking density by reacting with the chemical ingredients for vulcanization. This technique would lead to lower overall cure rates [34,35], and at the same time it degraded the mechanical performance of such silica filled material for tire applications. In summary, due to these flaws none of the methods mentioned above could simultaneously ensure both the ability in reducing the silica agglomeration and improving the material performance.

41
References [1] J. C. Brosse et al., J. Appl. Polym. Sci., 78, 1461 (2000). [2] A. F. Halasa et al., “Science and Technology of Rubber”, 2nd Ed., Academic Press, 1994. [3] D. Derouet, P. Phinyocheep, J. C. Brosse and G. Boccaccio, Eur. Polym. J., 26(12), 1301 (1990). [4] K. Chino, M. Ashiura, Macromolecules, 34, 9201 (2001). [5] F. Ferrero, M. Panetti and G. B. Saracco, La Chimica e L’Industria, 66, 3 (1984). [6] P. Dreyfuss, J. P. Kennedy, Anal. Chem., 47, 771 (1975). [7] A. Brydon, Ph.D. thesis, University of Aberdeen, 1972. [8] J. M. Stellmann, A. E. Woodward, J. Polym. Sci., A2, 52 (1971). [9] J. Malhorta et al., Polymer, 30, 467 (1989). [10] D. Zuchowska, Polymer, 21, 514 (1980). [11] A. Brydon et al., Makromol. Chem., 178, 1739 (1977). [12] J. March, “Advanced Organic Chemistry”, 4th Ed., Wiley, 1992. [13] R. C. Larock, “Comprehensive Organic Transformations”, 2nd Ed., Wiley, 1999. [14] BMBF project “Supramolekular strukturierte Elastomerkomposite mit adpativer Energiedissipation”, 2003.
![References [1] J. C. Brosse et al., J. Appl. Polym.](http://images.slideplayer.com/14/4235763/slides/slide_41.jpg "Sci., 78, 1461 (2000). [2] A. F. Halasa et al., Science and Technology of Rubber , 2nd Ed., Academic Press, [3] D. Derouet, P. Phinyocheep, J. C. Brosse and G. Boccaccio, Eur. Polym. J., 26(12), 1301 (1990). [4] K. Chino, M. Ashiura, Macromolecules, 34, 9201 (2001). [5] F. Ferrero, M. Panetti and G. B. Saracco, La Chimica e L’Industria, 66, 3 (1984). [6] P. Dreyfuss, J. P. Kennedy, Anal. Chem., 47, 771 (1975). [7] A. Brydon, Ph.D. thesis, University of Aberdeen, [8] J. M. Stellmann, A. E. Woodward, J. Polym. Sci., A2, 52 (1971). [9] J. Malhorta et al., Polymer, 30, 467 (1989). [10] D. Zuchowska, Polymer, 21, 514 (1980). [11] A. Brydon et al., Makromol. Chem., 178, 1739 (1977). [12] J. March, Advanced Organic Chemistry , 4th Ed., Wiley, [13] R. C. Larock, Comprehensive Organic Transformations , 2nd Ed., Wiley, [14] BMBF project Supramolekular strukturierte Elastomerkomposite mit adpativer Energiedissipation ,")
42
[15] K. Yurekli et al., J. Polym. Sci. Part B. Polym. Phys., 39 256 (2000). [16] H. Pawlowski and J. Dick, Rubber World, 6, 35 (1992). [17] F. W. Maine, B. E. Riseborough and J. E. Theberge, Polymer structures and properties, SPE RETEC, Toronto, 1976. [18] B. Freund, W. Niedermeier, Kautsch. Gummi Kunsts., 51, 444 (1998). [19] E. Guth and O. Gold, Phys. Rev., 53, 322 (1938). [20] H. M. Smallwood, J. Appl. Phys., 15, 758 (1944). [21] J. H. Davis, Plastics and Polymer, 39, 137 (1971). [22] J. Fröhlich and H. D. Luginsland, Rubber World, 4, 28 (2001). [23] J. D. Ferry, “Viscoelastic Properties of Polymers”, 3rd ed., Wiley, 1980. [24] G. M. Bartenev, “Structure and Relaxation Properties of Elastomers”, Khimiya, Moscow, 1979. [25] G. M. Bartenev, Doklady Akad. Nauk USSR, 300, 1154 (1988). [26] G. M. Bartenev, Vysokomol. Soed., A25, 1191 (1983). [27] D. F. Twiss, J.Chem. Soc., 44, 1067 (1925). [28] B. Meissner, Rubber Chem. Technol., 68, 297 (1995). [29] C. C. McCabe and N. Müller, Trans. Soc. Rheol., 5, 329 (1961). [30] J. L. White and J. W. Crowder, J. Appl. Polym. Sci., 18, 1013 (1974). [31] S. N. Maiti and P. K. Mahapatro, Polym. Compos., 9, 291 (1988). [32] G. I. Taylor, Proc. Rheo. Soc. London Ser. A, 146, 501 (1934). [33] N. Mills, J. Appl. Polym. Sci., 15, 2791 (1975). [34] F. A. Morrison, “Understanding Rheology”, Oxford University Press, 2001.
![[15] K. Yurekli et al., J. Polym. Sci. Part B.](http://images.slideplayer.com/14/4235763/slides/slide_42.jpg "Polym. Phys., (2000). [16] H. Pawlowski and J. Dick, Rubber World, 6, 35 (1992). [17] F. W. Maine, B. E. Riseborough and J. E. Theberge, Polymer structures and properties, SPE RETEC, Toronto, [18] B. Freund, W. Niedermeier, Kautsch. Gummi Kunsts., 51, 444 (1998). [19] E. Guth and O. Gold, Phys. Rev., 53, 322 (1938). [20] H. M. Smallwood, J. Appl. Phys., 15, 758 (1944). [21] J. H. Davis, Plastics and Polymer, 39, 137 (1971). [22] J. Fröhlich and H. D. Luginsland, Rubber World, 4, 28 (2001). [23] J. D. Ferry, Viscoelastic Properties of Polymers , 3rd ed., Wiley, [24] G. M. Bartenev, Structure and Relaxation Properties of Elastomers , Khimiya, Moscow, [25] G. M. Bartenev, Doklady Akad. Nauk USSR, 300, 1154 (1988). [26] G. M. Bartenev, Vysokomol. Soed., A25, 1191 (1983). [27] D. F. Twiss, J.Chem. Soc., 44, 1067 (1925). [28] B. Meissner, Rubber Chem. Technol., 68, 297 (1995). [29] C. C. McCabe and N. Müller, Trans. Soc. Rheol., 5, 329 (1961). [30] J. L. White and J. W. Crowder, J. Appl. Polym. Sci., 18, 1013 (1974). [31] S. N. Maiti and P. K. Mahapatro, Polym. Compos., 9, 291 (1988). [32] G. I. Taylor, Proc. Rheo. Soc. London Ser. A, 146, 501 (1934). [33] N. Mills, J. Appl. Polym. Sci., 15, 2791 (1975). [34] F. A. Morrison, Understanding Rheology , Oxford University Press,")
43
[35] Q. Zheng et al., J. Appl. Polym. Sci., 86, 3166 (2002). [36] D. Miao et al., Nihon Reoroji Gakkaishi, 31(5), 305 (2003). [37] Q. Zheng et al., Polymer, 42, 5743 (2001). [38] S. Vieweg et al., J. Appl. Polym. Sci., 73, 495 (1999). [39] V. Arrighi, I. J. McEwen, H. Qian and M. B. S. Prieto, Polymer, 44, 6259 [40] G. Tsagaropoulos and A. Eisenberg, Macromolecules, 28, 396 (1995). [41] G. Tsagaropoulos and A. Eisenberg, Macromolecules, 28, 6067 (1995). [42] S. Yano, T. Furukawa, M. Kodomari and K. Kurita, Kobunshi Rondunshu, 53, 218 (1996). [43] Y. I. Tien and K. H. Wei, J. Appl. Polym. Sci., 86, 1741 (2002). [44] Z. S. Petrovic and W. Zhang, Mater. Sci. Forum, 352, 171 (2000). [45] N. D. Alberola and P. Mele, Polymer Composites, 17, 751 (1996). [46] A. Yim, R. S. Chahal and L. E. St. Pierre, J. Colloid. Interface Sci., 43, 583 (1973). [47] C. J. T. Landry, B. K. Coltrain, M. R. Landry, J. J. Fitzgerald and V. K. Long, Macromolecules, 26, 3702 (1993). [48] M. Takayanagi, S. Uemura and S. Minami, J. Polym. Sci., C5, 113 (1968).
![[35] Q. Zheng et al., J. Appl. Polym. Sci., 86, 3166 (2002).](http://images.slideplayer.com/14/4235763/slides/slide_43.jpg "[36] D. Miao et al., Nihon Reoroji Gakkaishi, 31(5), 305 (2003). [37] Q. Zheng et al., Polymer, 42, 5743 (2001). [38] S. Vieweg et al., J. Appl. Polym. Sci., 73, 495 (1999). [39] V. Arrighi, I. J. McEwen, H. Qian and M. B. S. Prieto, Polymer, 44, 6259 [40] G. Tsagaropoulos and A. Eisenberg, Macromolecules, 28, 396 (1995). [41] G. Tsagaropoulos and A. Eisenberg, Macromolecules, 28, 6067 (1995). [42] S. Yano, T. Furukawa, M. Kodomari and K. Kurita, Kobunshi Rondunshu, 53, 218 (1996). [43] Y. I. Tien and K. H. Wei, J. Appl. Polym. Sci., 86, 1741 (2002). [44] Z. S. Petrovic and W. Zhang, Mater. Sci. Forum, 352, 171 (2000). [45] N. D. Alberola and P. Mele, Polymer Composites, 17, 751 (1996). [46] A. Yim, R. S. Chahal and L. E. St. Pierre, J. Colloid. Interface Sci., 43, 583 (1973). [47] C. J. T. Landry, B. K. Coltrain, M. R. Landry, J. J. Fitzgerald and V. K. Long, Macromolecules, 26, 3702 (1993). [48] M. Takayanagi, S. Uemura and S. Minami, J. Polym. Sci., C5, 113 (1968)..")
Similar presentations






 Date: April 14, 2000 Slide:1 Environmentally Conscious Design & Manufacturing Class 17: Plastics.>")








 – What color? Does it fluoresce.>")




 is.>")



