Download presentation
Presentation is loading. Please wait.

1
Hybridization and hypervalency
in transition-metal and main-group bonding (The Absence of) Radial Nodes of Atomic Orbitals, an Important Aspect in Bonding Throughout the Periodic Table Consequences for p-Block Elements: Hybridization Defects, Hypervalency, Bond Polarity, etc…. Consequences für the Transition Metal Elements Non-VSEPR Structures of d0 Complexes Martin Kaupp, Winter School on Quantum Chemistry, Helsinki, Dec , 2009
 Radial Nodes of Atomic Orbitals, an Important Aspect in Bonding Throughout the Periodic Table. Consequences for p-Block Elements: Hybridization Defects, Hypervalency, Bond Polarity, etc…. Consequences für the Transition Metal Elements. Non-VSEPR Structures of d0 Complexes. Martin Kaupp, Winter School on Quantum Chemistry, Helsinki, Dec ,")
2
Analogy between Wave Functions and Classical Standing Waves
Vibrations of a string fixed at both ends. Base tone and the first two harmonics are shown. Note the nodes (zero crossings)!! They result from the necessary orthogonality of the modes. 2. harmonic 1. harmonic x=0 x=l base tone
!! They result from the necessary. orthogonality of the modes. 2. harmonic. 1. harmonic. x=0. x=l. base tone.")
3
The Nodes of the Radial Solutions
R is the product of an exponential e-(1/2)r and of an associated Laguerre polynomial; r = 2Zr/na0 1s, 2p, 3d, 4f: no „primogenic repulsion“ (Pekka Pyykkö)
r and of an. associated Laguerre polynomial; r = 2Zr/na0. 1s, 2p, 3d, 4f: no „primogenic repulsion (Pekka Pyykkö)")
4
1s 2p 2p 3d 4f See also: J. Comput. Chem. 2007, 28, 320.

5
P-Block Main-Group Elements
See also: J. Comput. Chem. 2007, 28, 320. P-Block Main-Group Elements

6
Hybridization Defects and their Consequences in p-Block Chemistry

7
and of „Saturated“ Hydrides MHn
X-H Binding Energies of Monohydrides XH, and of „Saturated“ Hydrides MHn W. Kutzelnigg Angew. Chem. 1984, 96, 262.

8
Radial Extent and Energies of Valence Orbitals
W. Kutzelnigg Angew. Chem. 1984, 96, 262. AO-Energies from Moore Tables 26-55% 20-33% 24-40% 10% r-expectation values from Desclaux tables (DHF)
")
9
Reasons for Hybridization (1)
better bonding overlap (radial) reduced antibonding overlap (angular)
 reduced antibonding overlap. (angular)")
10
Reasons for Hybridization (2)
W. Kutzelnigg, Einführung in die Theoretische Chemie, Vol. 2

11
Hybridization Defects (1)
Mulliken gross populations for valence AOs in XHn hydrides without free electron pairs W. Kutzelnigg Angew. Chem. 1984, 96, 262.

12
CH4 sp3.16 SiH4 sp2.10 GeH4 sp2.00 SnH4 sp1.81 PbH4 sp1.77
Hybridization Defects (2) cos(180-) = a2/(1-a2) (a2 = s-character, 1-a2 = p-character) Holds only for orthogonal hybrids! NPA/NLMO Hybridization* CH4 sp3.16 SiH4 sp2.10 GeH4 sp2.00 SnH4 sp1.81 PbH4 sp1.77 *A posteriori hybridization analyses from DFT calculations (BP86/TZVP)
 cos(180-) = a2/(1-a2) (a2 = s-character, 1-a2 = p-character) Holds only for orthogonal hybrids! NPA/NLMO Hybridization* CH4. sp3.16. SiH4. sp2.10. GeH4. sp2.00. SnH4. sp1.81. PbH4. sp1.77. *A posteriori hybridization analyses from DFT calculations (BP86/TZVP)")
13
Explanations of the Inert-Pair Effect
1) Promotion energies increase down the group 2) Drago: Binding energies decrease down the group promotion energies are not overcompensated anymore 3) Extension following Kutzelnigg: Promotion does not take place anymore no good hybrids and weakened covalent bonds in higher oxidation state
 Promotion energies increase down the group. 2) Drago: Binding energies decrease down the group. promotion energies are not overcompensated anymore. 3) Extension following Kutzelnigg: Promotion does not take place anymore no good hybrids and weakened covalent bonds in. higher oxidation state.")
14
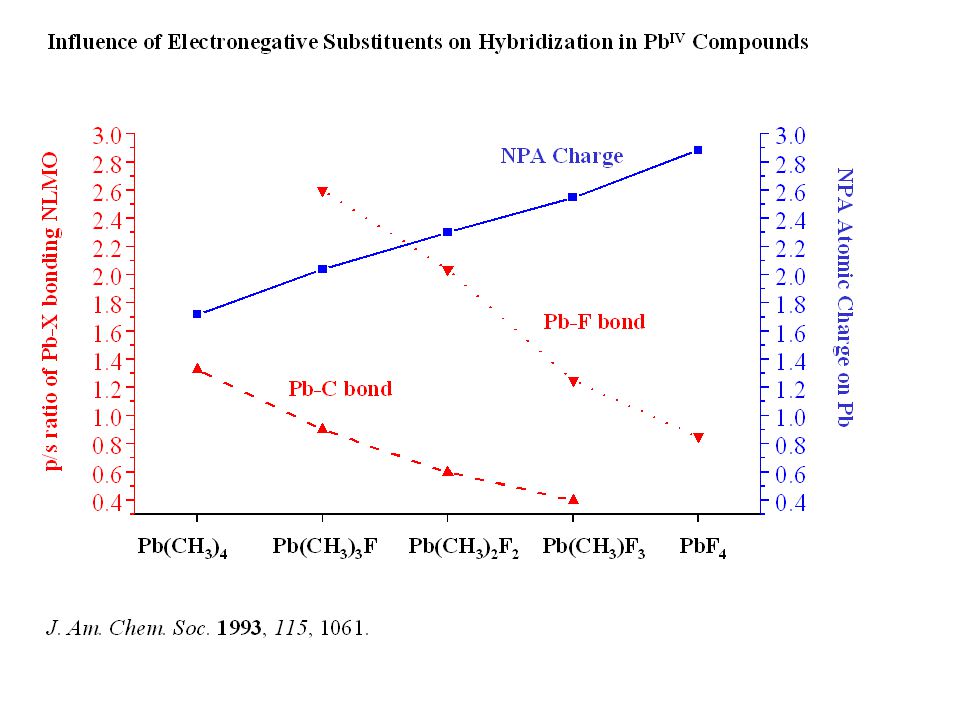
inorganic lead chemistry:
Why is Inorganic Lead Chemistry Dominated by PbII, but Organolead Chemistry by PbIV? (J. Am. Chem. Soc. 1993, 115, 1061) Pb(IV) Pb(II) inorganic lead chemistry: PbCl4: decomposes at low temperatures PbCl2, PbO, Pb(OAc)2: stable ionic compounds PbO2 or Pb(OAc)4: strong oxidation agents organolead chemistry: PbMe4, PbEt4: stable compounds, earlier appreciable commercial value PbMe2, PbEt2 unknown, possibly reactive intermediates decreasing stability: R4Pb > R3PbX > R2PbX2 > RPbX3 > PbX4 (R = alkyl, aryl; X = halogen, OR, NR2, OOCR, etc……)
 Pb(IV) Pb(II) inorganic lead chemistry: PbCl4: decomposes at low temperatures. PbCl2, PbO, Pb(OAc)2: stable ionic compounds. PbO2 or Pb(OAc)4: strong oxidation agents. organolead chemistry: PbMe4, PbEt4: stable compounds, earlier appreciable commercial value. PbMe2, PbEt2. unknown, possibly reactive intermediates. decreasing stability: R4Pb > R3PbX > R2PbX2 > RPbX3 > PbX4. (R = alkyl, aryl; X = halogen, OR, NR2, OOCR, etc……)")
15
) (kJ mol E D QCISD/DZ+P data - 60 40 20 80 100 120 140 160 PbF
80 100 120 140 160 PbF 4 PbMeF 3 PbMe 2 F n +F 1 +MeF +C H 6 D E react. (kJ mol ) Substituent Effects on 1,1 Elimination Reactions of Pb(IV) Compounds J. Am. Chem. Soc. 1993 , 115 , 1061. QCISD/DZ+P data
 Substituent Effects on 1,1. Elimination Reactions of Pb(IV) Compounds. J. Am. Chem. Soc , 115. , QCISD/DZ+P data.")
16
Substituent Effects on 1,1-Elimination Reactions of Pb(IV) Compounds
PbMenF4-n→ PbMenF2-n +F2 -60 -40 -20 20 40 60 80 100 120 140 160 DEreact. (kJ mol-1) PbMenF4-n→ PbMen-1F3-n +MeF PbMenF4-n→ PbMen-2F4-n +C2H6 PbF4 PbMeF3 PbMe2F2 PbMe3F PbMe4 J. Am. Chem. Soc. 1993, 115, 1061. QCISD/DZ+P data
 PbMenF4-n→ PbMen-1F3-n +MeF. PbMenF4-n→ PbMen-2F4-n +C2H6. PbF4. PbMeF3. PbMe2F2. PbMe3F. PbMe4. J. Am. Chem. Soc. 1993, 115, QCISD/DZ+P data.")
17
sp3.16 sp1.98 sp2.10 sp1.01 sp2.00 sp0.75 sp1.81 sp0.70 sp1.77 sp0.51
Hybridization Defects (3) EH4 h(E) charge Q(E) EF4 CH4 sp3.16 -0.952 CF4 sp1.98 1.338 SiH4 sp2.10 0.547 SiF4 sp1.01 2.361 GeH4 sp2.00 0.392 GeF4 sp0.75 1.852 SnH4 sp1.81 0.753 SnF4 sp0.70 2.348 PbH4 sp1.77 0.519 PbF4 sp0.51 2.111 h(E): NPA/NLMO Hybridization results from DFT calculations (BP86)
 EH4. h(E) charge Q(E) EF4. CH4. sp CF4. sp SiH4. sp SiF4. sp GeH4. sp GeF4. sp SnH4. sp SnF4. sp PbH4. sp PbF4. sp h(E): NPA/NLMO Hybridization. results from DFT calculations (BP86)")
19
Bent‘s Rule: Basic Principles
e.g.: larger angles between more electropositive substituents more s-character of central atom is directed towards less electronegative substituent, more p-character to more electronegative substituent H. A. Bent Chem. Rev. 1961, 61, 275; H. C. Allen at al. J. Am. Chem. Soc. 1958, 80, 2673. exceptions: a) large steric effects, b) very different sizes of substituent orbitals (e.g. H vs. Hg), c) counteracting effects for very light central atoms d) transition metal systems (cf. also Huheey text book)
 large steric effects, b) very different sizes of substituent orbitals (e.g. H vs. Hg), c) counteracting effects for very light central atoms d) transition metal systems (cf. also Huheey text book)")
20
Bent‘s rule effects in organolead fluorides
101.10 Pb 115.50 Pb 116.40 102.80 101.40 104.10 Pb 134.80 Quasirelativistic ab initio calculations, J. Am.Chem. Soc. 1993, 115, 1061.

21
Bent‘s Rule: Counteracting Effects
negative hyperconjugation „switched off“ negative hyperconjugation counteracts EN effects negative hyperconjugation small for heavier elements

22
EH3 h(E-H) h(LP-E) NH3 sp3.14 sp2.61 PH3 sp5.29 sp0.76 AsH3 sp7.08
Bent‘s Rule and Lone Pairs EH3 h(E-H) h(LP-E) NH3 sp3.14 sp2.61 PH3 sp5.29 sp0.76 AsH3 sp7.08 sp0.51 h: NPA/NLMO Hybridization results from DFT calculations (BP86)
 h(LP-E) NH3. sp3.14. sp2.61. PH3. sp5.29. sp0.76. AsH3. sp7.08. sp0.51. h: NPA/NLMO Hybridization. results from DFT calculations (BP86)")
23
Inversion Barriers and Hybridization
† TS pure p-character of LP high s-character in bonds high s-character of LP high p-character in bonds everything that enhances hybridization defects increases inversion barrier and decreases angles! e.g. NH3 << PH3 < AsH3 < SbH3 < BiH3 NLi3 << NH3 << NF3 NH3 << NH2F << NHF2 << NF3

24
Further Pecularities of 1st-Row (2. Period) Elements (p-Block)
important: size, electronegativity, hybridization defects particularly weak bonds for F2, H2O2, N2H4, e.g. BDE (kJ mol‑1): F2 155, Cl2 240, Br2 190, I2 149. (cf. also low EA of fluorine atom!) explanation: „lone-pair bond weakening effect“ (Sanderson), (less pronounced for heavier homologues) preference for - multiple bonding compared to chain-linked single bonds e.g.: O2 vs S8, CO2 vs. SiO2, ketones vs. silicones e.g. BDE (kJ mol‑1): N-N P-P single 167 200 double 418 310 triple 946 490
: F2 155, Cl2 240, Br2 190, I (cf. also low EA of fluorine atom!) explanation: „lone-pair bond weakening effect (Sanderson), (less pronounced for heavier homologues) preference for - multiple bonding compared to chain-linked single bonds. e.g.: O2 vs S8, CO2 vs. SiO2, ketones vs. silicones. e.g. BDE (kJ mol‑1): N-N. P-P. single double triple")
25
Binding Energies

26
234 213 I 310 285 Br 381 327 Cl 565 485 F Si-X C-X X BDEs (kJ mol‑1):
Preference for - multiple bonding compared to chain-linked single bonds increments (/, in kJ mol‑1): C-C N-N O-O 322/295 160/395 145/350 Si-Si P-P S-S 195/120 200/145 270/155 Ge-Ge As-As Se-Se 165/110 175/120 210/125 also: bond ionicity cf. silicates, etc…! 234 213 I 310 285 Br 381 327 Cl 565 485 F Si-X C-X X BDEs (kJ mol‑1):
: C-C. N-N. O-O. 322/ / /350. Si-Si. P-P. S-S. 195/ / /155. Ge-Ge. As-As. Se-Se. 165/ / /125. also: bond ionicity. cf. silicates, etc…! I Br Cl F. Si-X. C-X. X. BDEs (kJ mol‑1):")
27
Pauling Allred-Rochow

28
Silicon-Oxygen Bonds 111o 154o
Electronegativity Si: O: 3.50 111o 154o Electron-Localisation-Function Electron-Localisation-Function The Si-O Bond has a Strong Ionic Character

29
Summary: Hybridization Defects and Related Phenomena in p-Block Main-Group Chemistry
2p shell has no primogenic repulsion and is thus compact this favors isovalent hybridization in the 2nd period electronegative substituents enhance hybridization defects for the heavier p-block elements, hybridization defects are important for inert-pair effect, inversion barriers, Bent‘s rule effects, double-bond rule….. large electronegativity of 2p-elements also arises from compact 2p shell

30
Hypervalency? On Myth and Reality of d-Orbital-Participation in Main-Group Chemistry

31
MgF64- SiF62- SF6 covalency increases ionicity increases
On the Definition of Hyper-(co)-Valency MgF SiF SF6 covalency increases ionicity increases certainly no hypervalency hypervalency ?
-Valency. MgF64- SiF62- SF6. covalency increases. ionicity increases. certainly no. hypervalency. hypervalency")
32
A Working Definition of Hypervalency
We regard a molecule as hypervalent if at least for one atom the bonding situation requires more valence orbitals than available for the free atom

33
The Early History of Hypervalency, Part 1
electron pair theory, octet rule Lewis 1916; Kossel 1916; Langmuir 1919 directed valency, spmdn hybrids, electroneutrality principle Pauling 1931, 1940, 1948 3-center-4-electron-bond Pimentel 1951; Hach, Rundle 1951

34
F Xe F F - Xe+ F F Xe + F - F - Xe2+ F - covalent, hypervalent
Limiting Bonding Situations: Example of Different Lewis Structures for XeF2 F - Xe2+ F - F Xe F F Xe + F - F - Xe+ F covalent, hypervalent partially ionic, delocalized ionic

35
3-Center-4-Electron MO Model
F Xe + F - F - Xe+ F sa sb sn pa pb pn

36
Modified 3-Center-4-Electron MO Model
F Xe + F - F - Xe+ F sa sb sn pa pb pn

37
The Early History of Hypervalency, Part 2
discovery of noble-gas compounds Chernik et al.; Claasen et al.; Bartlett, Hoppe; since 1962: first ab initio calculations since ca. 1970 first improved wave functions, Mulliken population analyses since ca. 1975:

38
+ l = pp + l dp = polarized pp The Role of d-Polarization-Functions
electric field

39
weak points of Mulliken population analysis:
strong basis-set dependence unphysical (partly negative) populations failure for transition to ionic bonding
 populations. failure for transition to ionic bonding.")
40
Improved Analyses of Accurate Wave Functions
modified Roby/Davidson population analysis Ahlrichs et al. 1976, 1985; Wallmeier, Kutzelnigg 1979 „Natural Population Analysis“ (NPA), „Natural Bond Orbital Analysis“ (NBO) Reed, Weinhold 1986; Reed, Schleyer 1990 „Natural Resonance Theory“ (NRT) Weinhold et al., 1995 energy contributions from d-orbitals Magnusson 1986, 1990, 1993 topological analysis of electron density (QTAIM) Cioslowski, Mixon 1993 „Generalized Valence Bond“ (GVB) theory Hay 1977, Cooper et al (full-GVB) one-center expansion of one-particle density matrix Häser 1996 Since late 1990‘s QTAIM based on experimental densities (e.g. D. Stalke et al.)
, „Natural Bond Orbital Analysis (NBO) Reed, Weinhold 1986; Reed, Schleyer „Natural Resonance Theory (NRT) Weinhold et al., energy contributions from d-orbitals. Magnusson 1986, 1990, topological analysis of electron density (QTAIM) Cioslowski, Mixon „Generalized Valence Bond (GVB) theory. Hay 1977, Cooper et al (full-GVB) one-center expansion of one-particle density matrix. Häser Since late 1990‘s. QTAIM based on experimental densities. (e.g. D. Stalke et al.)")
41
- O S O - - O S2+ O - - O S2+ O - NRT-Analysis of Sulfate Ion
NRT weight: O 0% - O S O - O O - - O S2+ O - 66.2% O - O - O S2+ O - 12x1.9% = 22.8% O 2- 11% NRT at HF/6-31G* level Weinhold et al., 1995 further ionic structures:

42
- O Cl3+ O - - O Cl3+ O - 0% 60.9% 12x2.0% = 24.0% 15% O O Cl O - O
Which NRT-Lewis Structure Describes the Perchlorate Ion Best? NRT weight: O 0% O Cl O - O O - - O Cl3+ O - 60.9% O - O - O Cl3+ O - 12x2.0% = 24.0% O 2- further ionic structures: 15% NRT at HF/6-31G* level Weinhold et al., 1995

43
H C C C C C H p3* su Hyperconjugation Exemplified by Cyclopentadiene
...... H C C p3* su C C C H

44
F F C H H p(F) s*(C-F) F F- F+ F+ F- C C C H H H
Negative Hyperconjugation Exemplified by Difluoromethane C H F C H F- F+ C H F+ F- ...... p(F) F F s*(C-F) C H H
 F. F. s*(C-F) C. H. H.")
45
P F O p(O) s*(P-F) (to e-MO) (to e*-MO) F- F F
Negative Hyperconjugation in F3PO F F- F P+ O- P+ O P+ O F F F F- ...... F F P F O p(O) (to e-MO) s*(P-F) (to e*-MO)
 (to e-MO) s*(P-F) (to e*-MO)")
46
…….and for p-backbonding in phosphane complexes…….
p(M) s*(P-F) P F M
 s*(P-F) P. F. M.")
47
e.g. XeF2, XeF4, XeF6, SF4, ClF3, etc….
Musher’s Classification of „Hypervalent“ Compounds (Musher 1969) 1. order 2. order e.g. XeF2, XeF4, XeF6, SF4, ClF3, etc…. lone pair present s-character concentrates in LP e.g. PF5, SF6, JF7 highest possible oxidation state s-character in bonds
 1. order. 2. order. e.g. XeF2, XeF4, XeF6, SF4, ClF3, etc…. lone pair present. s-character. concentrates in LP. e.g. PF5, SF6, JF7. highest possible. oxidation state. s-character in bonds.")
48
MO-Diagram of SF6

49
Summary: Hypervalency, d-Orbital-Participation in p-Block Main-Group Chemistry
in normal valent and „hypervalent“ p-block main-group systems, d-functions serve as „polarization functions“ but not as true valence orbitals hypervalent Lewis structures usually obtain small or no weight! ionic bonding contributions dominate, assisted by delocalization (3-c-4-el.-bonding, negative hyperconjugation) in PF5, SF6, etc., delocalization is more complex due to s-orbital participation significant hypervalent populations
 in PF5, SF6, etc., delocalization is more complex due to s-orbital participation significant hypervalent populations.")
50
Transition Metal Systems
3d

51
Radial Orbital Densities of the Manganese Atom
r f(r) in a.u. r in a.u. J. Comput. Chem. 2007, 28, 320
 in a.u. r in a.u. J. Comput. Chem. 2007, 28, 320.")
52
M L (n-1)d (n-1)s,p Pauli repulsion outermost stretched bond
The problem of the outermost core shells in transition-metal atoms M L (n-1)s,p outermost core shell Pauli repulsion stretched bond (n-1)d The problem is most pronounced for the 3d elements (nodelessness of the 3d shell) weak bonds, low-lying excited states, strong (nondynamical) correlation effects The 4d shell is already better suited for bonding overlap. The 5d shell even more so due to its relativistic expansion. M. Kaupp J. Comput. Chem. 2007, 28, 320. See also: M. A. Buijse, E. J. Baerends J. Chem. Phys. 1990, 93, 4129.
s,p. outermost. core shell. Pauli repulsion. stretched bond. (n-1)d. The problem is most pronounced for the 3d elements. (nodelessness of the 3d shell) weak bonds, low-lying excited states, strong (nondynamical) correlation effects. The 4d shell is already better suited for bonding overlap. The 5d shell even more so due to its relativistic expansion. M. Kaupp J. Comput. Chem. 2007, 28, 320. See also: M. A. Buijse, E. J. Baerends J. Chem. Phys. 1990, 93,")
53
M (n-1)d (n-1)s,p Pauli repulsion outermost stretched bond
The problem of the outermost core shells in transition-metal atoms M (n-1)s,p outermost core shell Pauli repulsion stretched bond (n-1)d The problem is most pronounced for the 3d elements (nodelessness of the 3d shell) weak bonds, low-lying excited states, strong (nondynamical) correlation effects The 4d shell is already better suited for bonding overlap. The 5d shell even more so due to its relativistic expansion. M. Kaupp J. Comput. Chem. 2007, 28, 320. See also: M. A. Buijse, E. J. Baerends J. Chem. Phys. 1990, 93, 4129.
s,p. outermost. core shell. Pauli repulsion. stretched bond. (n-1)d. The problem is most pronounced for the 3d elements. (nodelessness of the 3d shell) weak bonds, low-lying excited states, strong (nondynamical) correlation effects. The 4d shell is already better suited for bonding overlap. The 5d shell even more so due to its relativistic expansion. M. Kaupp J. Comput. Chem. 2007, 28, 320. See also: M. A. Buijse, E. J. Baerends J. Chem. Phys. 1990, 93,")
54
M L (n-1)d (n-1)s,p Pauli repulsion outermost stretched bond
The problem of the outermost core shells in transition-metal atoms M L (n-1)s,p outermost core shell Pauli repulsion stretched bond (n-1)d The problem is most pronounced for the 3d elements (nodelessness of the 3d shell) weak bonds, low-lying excited states, strong (nondynamical) correlation effects The 4d shell is already better suited for bonding overlap. The 5d shell even more so due to its relativistic expansion. M. Kaupp J. Comput. Chem. 2007, 28, 320. See also: M. A. Buijse, E. J. Baerends J. Chem. Phys. 1990, 93, 4129.
s,p. outermost. core shell. Pauli repulsion. stretched bond. (n-1)d. The problem is most pronounced for the 3d elements. (nodelessness of the 3d shell) weak bonds, low-lying excited states, strong (nondynamical) correlation effects. The 4d shell is already better suited for bonding overlap. The 5d shell even more so due to its relativistic expansion. M. Kaupp J. Comput. Chem. 2007, 28, 320. See also: M. A. Buijse, E. J. Baerends J. Chem. Phys. 1990, 93,")
55
Non-VSEPR Structures and Bonding for d0-Systems

56
Non-VSEPR Structures of d0-Systems
VSEPR = valence-shell electron-pair-repulsion model

58
Practical Relevance of d0-Systems
e.g. catalysis e.g. bioinorganic chemistry ZrO2, ferroelectric perovskites e.g. materials Mo-Cofactor (after Rajagopalan, Johnson)
")
59
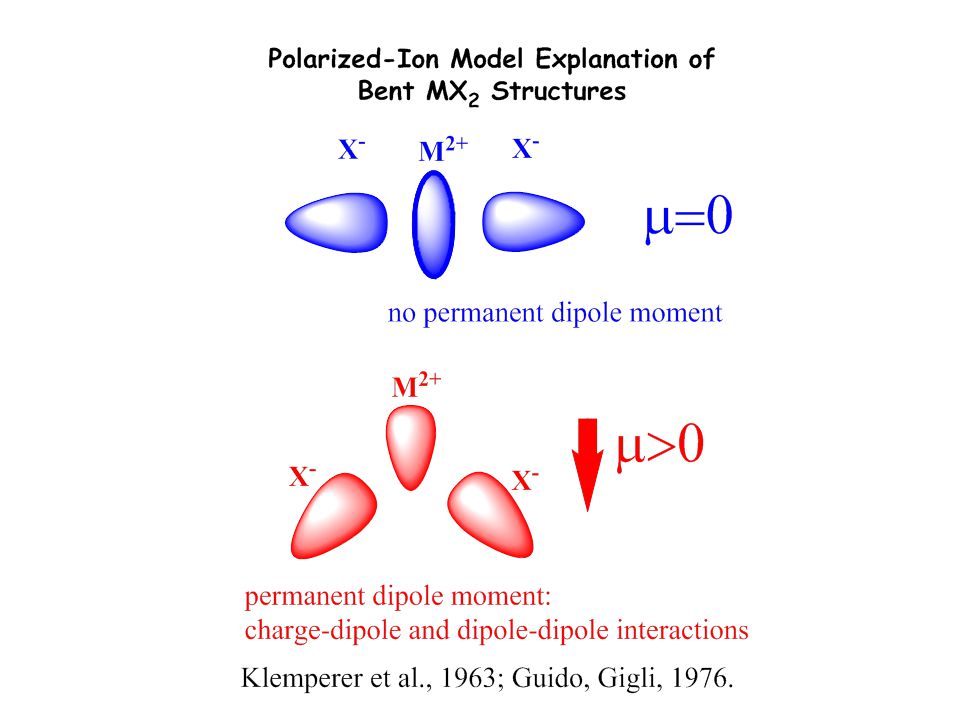
The Question of Bending
Mg F The Question of Bending of the Alkaline Earth Dihalides 1230 Ba F linearization energies in kJ/mol from SDCI calculations (J. Am. Chem. Soc. 1991, 113, 6012)
")
60
M X Factors Controlling the Structures of d0 Systems
maximization of d-orbital participation in -bonding mutual repulsion and polarization of the ligands distorted, „non-VSEPR“ structures regular, „VSEPR“ structures polarization of central-atom semi-core orbitals by the ligands, „inverse polarization“ of cation maximization of -bonding M X (??) Review: Angew. Chemie 2001, 113, 642.
 Review: Angew. Chemie 2001, 113, 642.")
61
d-Orbital-Contributions to HOMO in d0 MX2-Complexes

63
projected on electron density (cloud of points)
BaH2: Electron Localization Function (ELF, color scale) projected on electron density (cloud of points)
 projected on electron density (cloud of points)")
64
Bending Force Constants (at Linear Structure)
for Molecular Alkaline Earth Dihalides J. Am. Chem. Soc. 1991, 113, 6017 force constants from polarized ion model too low for Be and Mg complexes but too high for Ca, Sr, and Ba complexes

65
Radial Distribution of (Pseudo)-Valence Orbitals of Barium
0.20 5p 0.15 6p 5d 0.10 r f(r) (a.u.) 0.05 6s 0.00 -0.05 2 4 6 r (Å) outer-core polarization and d-orbital participation cannot be separated completely!
 (a.u.) s r (Å) outer-core polarization and d-orbital participation cannot be separated completely!")
66
M X Factors Controlling the Structures of d0 Systems
maximization of d-orbital participation in -bonding mutual repulsion and polarization of the ligands distorted, „non-VSEPR“ structures regular, „VSEPR“ structures polarization of central-atom semi-core orbitals by the ligands, „inverse polarization“ of cation maximization of -bonding M X (??) Review: Angew. Chemie 2001, 113, 642.
 Review: Angew. Chemie 2001, 113, 642.")
67
Low Ligand-Ligand Repulsion for Neutral Ligands
J. Phys. Chem. 1992, 96, 7316 Ba2+(H2O)2 Ba DE(MP2)Lin= 0.6 kJ/Mol O O 1170 Ba2+(H2O)3 Ba O O O DE(HF)Plan= 0.4 kJ/Mol 980
2. Ba. DE(MP2)Lin= 0.6 kJ/Mol. O. O Ba2+(H2O)3. Ba. O. O. O. DE(HF)Plan= 0.4 kJ/Mol")
68
Cs+(H2O)2 Cs O O 1220 DE(MP2)Lin= 0.2 kJ/Mol
2 Cs O O 1220 DE(MP2)Lin= 0.2 kJ/Mol")
69
M X Factors Controlling the Structures of d0 Systems
maximization of d-orbital participation in -bonding mutual repulsion and polarization of the ligands distorted, „non-VSEPR“ structures regular, „VSEPR“ structures polarization of central-atom semi-core orbitals by the ligands, „inverse polarization“ of cation maximization of -bonding M X (??) Review: Angew. Chemie 2001, 113, 642.
 Review: Angew. Chemie 2001, 113, 642.")
70
X: Li BeH BH2 CH3 NH2 OH F C2v(in-plane) Cs C2v(out-of-plane) Ba Ba
Effects of p-Bonding C2v(in-plane) X: Li BeH BH2 CH3 NH2 OH F 5 10 15 20 25 30 DElin/ kJ mol-1 Cs C2v(out-of-plane) Ba Ba N N N N 118.4º 128.8º C2v(in-plane) DE(HF) = 0.0 kJ mol-1 C2v(out-of-plane) DE(HF) = kJ mol-1
 X: Li BeH BH2 CH3 NH2 OH F DElin/ kJ mol-1. Cs. C2v(out-of-plane) Ba. Ba. N. N. N. N º º. C2v(in-plane) DE(HF) = 0.0 kJ mol-1. C2v(out-of-plane) DE(HF) = kJ mol-1.")
71
MR2 (M = Ca, Yb, Eu; R = C[Si(CH3)3]3 )
Eaborn et al. Organomet. 1996, 15, 4783. J. Chem. Soc., Chem. Commun. 1997, 1961.
![MR2 (M = Ca, Yb, Eu; R = C[Si(CH3)3]3 )](http://slideplayer.com/slide/4181107/14/images/71/MR2+%28M+%3D+Ca%2C+Yb%2C+Eu%3B+R+%3D+C%5BSi%28CH3%293%5D3+%29.jpg "Eaborn et al. Organomet. 1996, 15, J. Chem. Soc., Chem. Commun. 1997,")
72
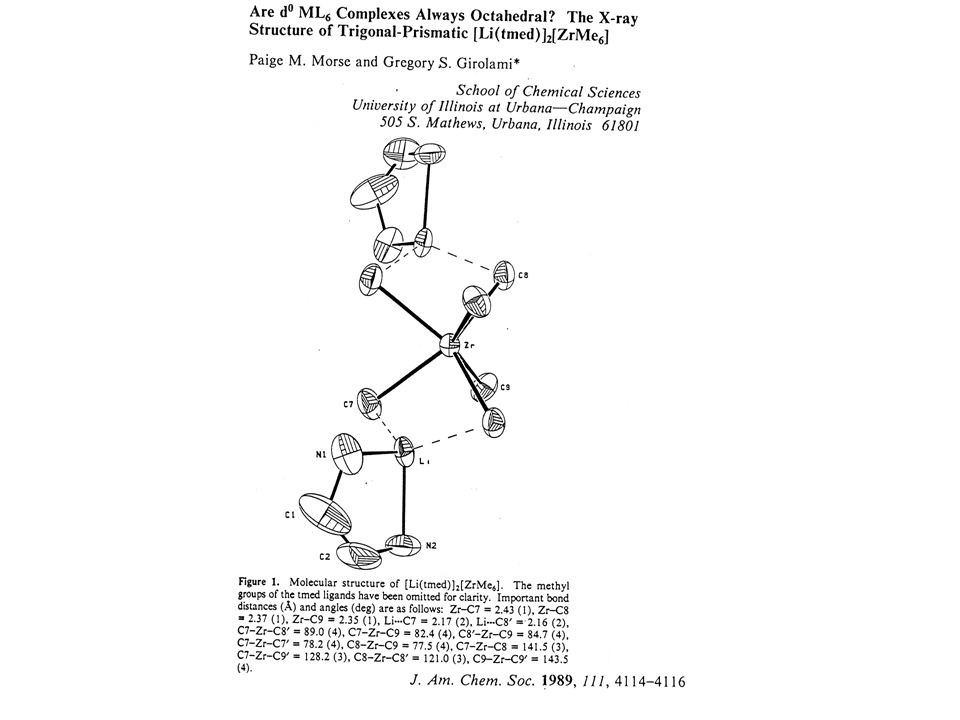
Sc H H H F Sc F F Structural Preferences of d0 MX3 Complexes
C. A. Jolly, D. S. Marynick Inorg. Chem. 1989, 28, 2893.

73
p-Acceptor-Type d-Orbitals for Linear and Bent MX2 d0 Complexes
‘pip’, ‘b2’ ‘pop’, ‘a2’ X ‘pip’, a1 ‘pop’, a2 M X X ‘pop’, b1

74
p-Bonding Favors Bent Structure in some Cases!

75
p-Bonding Favors Bent Structure in some Cases!

76
Cl Cl Ti Si C C (CH3)2SiCl2 (CH3)2TiCl2
Inversion of Bent’s rule for d0-Systems Ti C Cl 117.30 102.80 Si C Cl 107.50 114.20 (CH3)2SiCl2 (CH3)2TiCl2 GED data, Haaland et al., 1996. Explanation via p-contributions: Chem. Eur. J. 1999, 5, 3632.
2SiCl2. (CH3)2TiCl2. GED data, Haaland et al., Explanation via p-contributions: Chem. Eur. J. 1999, 5,")
78
Examples for Nonoctahedral Complexes with Sulfur Ligands
typical ligand types: See, e.g.: Can. J. Chem. 1989, 67, 1914 Inorg. Chem. 1993, 32, 2085 Inorg. Chem. 1989, 28, 773, etc.....

80
95.60 2.15 Å 2. 18 Å 2.21 Å 75.60 84.60 The Structure of W(CH3)6 W W
D3 C3 transition state minimum +17 kJ/mol CCSD(T) M. Kaupp J. Am. Chem. Soc. 1996, 118, 3018.
 M. Kaupp J. Am. Chem. Soc. 1996, 118,")
82
MO Rationalization of Nonoctahedral d0 Structures
S. K. Kang, H. Tang, T. A. Albright J. Am. Chem. Soc. 1993, 115, 1971.

83
Landis’ sd-Hybridization Model (1)
(J. Am. Chem. Soc. 1998, 120, 1842)
")
84
Landis’ sd-Hybridization Model (2)
(J. Am. Chem. Soc. 1998, 120, 1842) Preferred coordination arrangements for sd5 hybrids.
 Preferred coordination arrangements for sd5 hybrids.")
85
Energetics of Baylar Twist for MX6 d0 Complexes
D3h M M transition state Oh minimum Relative energies (kJ mol—1) between D3h and Oh structures of group 6 hexahalide complexes Cr Mo W F 61.6 27.1 42.9 Cl 56.4 62.6 78.7 Br -a 61.9 74.9 I 66.4 aDFT-BP86 results, Angew. Chem., Int. Ed. Engl. 2001, 40, 3534. No convergence for CrBr6, CrI6, MoI6.
 between D3h and Oh. structures of group 6 hexahalide complexes. Cr. Mo. W. F Cl Br. -a I aDFT-BP86 results, Angew. Chem., Int. Ed. Engl. 2001, 40, No convergence for CrBr6, CrI6, MoI6.")
86
and trigonal prismatic
dihedral d = 26º 2.414 Å 88.7º 2.406 Å 83.5º Mo S 5 10 15 20 25 30 35 40 45 50 55 60 Mo(SH)6: intermediate between octahedral and trigonal prismatic Erel / kJ mol-1 d / deg
6: intermediate. between octahedral. and trigonal prismatic. Erel / kJ mol-1. d / deg.")
87
of certain molybdenum enzymes
Importance of trigonal prismatic intermediates and transition structures of certain molybdenum enzymes Review: M. Kaupp Angew. Chemie, Int. Ed. Engl. 2004, 43, 546.

88
The Structure of WCl5CH3 83.30 79.90 C Cl3 2.19 Å 2.32 Å Cl2 78.10 C Cl4 102.10 2.35 Å W 2.22 Å Cl2 Cl1 81.10 Cl3 W 2.39 Å 2.33 Å 75.60 2.34 Å 81.90 2.35 Å 2.35 Å Cl5 Cl1 Cl5 Cl4 85.30 <Cl1-W-Cl5 = 97.90 <Cl1-W-Cl2 = <Cl3-W-Cl4 = <Cl1-W-Cl3 = 87.00 Cs oct. Cs trig. prism transition state minimum +19 kJ/mol BP86/A BP86 data, relative energies in kJ/mol Angew. Chemie 1999, 111, 3219

89
tp “same face”, Cs (TS +14.5) cis oct. C2v (TS +25.7)
The Structure of WCl4(CH3)2 tp “same face”, Cs (TS +14.5) cis oct. C2v (TS +25.7) cis tp “other face”, C2v (TS +8.5) trans oct. C2 (min. 0.0) “intermediate” C2 (min. +6.5) BP86 data, relative energies in kJ/mol Angew. Chemie 1999, 111, 3219
2. tp same face , Cs. (TS +14.5) cis oct. C2v. (TS +25.7) cis tp other face , C2v. (TS +8.5) trans oct. C2. (min. 0.0) intermediate C2. (min. +6.5) BP86 data, relative energies in kJ/mol. Angew. Chemie 1999, 111,")
90
oct. fac. C3v (TS +61.2) oct. merid. Cs (TS +26.0) tp “across” C1
The Structure of WCl3(CH3)3 oct. fac. C3v (TS +61.2) oct. merid. Cs (TS +26.0) tp “across” C1 (min. 0.0) tp “same face” C3 (min. +2.3) BP86 data, relative energies in kJ/mol Angew. Chemie 1999, 111, 3219
3. oct. fac. C3v. (TS +61.2) oct. merid. Cs. (TS +26.0) tp across C1. (min. 0.0) tp same face C3. (min. +2.3) BP86 data, relative energies in kJ/mol. Angew. Chemie 1999, 111,")
91
Examples of Unsymmetrical Metal Coordination in Oxide Materials oxide
related material properties ZrO2 7-fold coordination (baddeleyite) vs. cubic high-temperature form refractive ceramics (stabilization of cubic form) WO3 6-fold, dist. octahedral electrochromic ceramics MoO3 6-fold, strongly dist. octahedral heterogeneous catalyst BaTiO3, etc.* ferroelectric and piezoelectric ceramics *5d vs. 4d comparisons: KTaO3 is regular octahedral (cubic) and paraelectric, KNbO3 is distorted (rhombohedral) and ferroelectric at lower temperatures. LiTaO3 has a ferroelectric transition at lower temperature (950 K) than LiNbO3 (1480 K). More ionic bonding and larger band gap with 5d vs. 4d, due to relativistic effects!
 vs. cubic high-temperature form. refractive ceramics. (stabilization of cubic form) WO3. 6-fold, dist. octahedral. electrochromic ceramics. MoO3. 6-fold, strongly dist. octahedral. heterogeneous catalyst. BaTiO3, etc.* ferroelectric and piezoelectric ceramics. *5d vs. 4d comparisons: KTaO3 is regular octahedral (cubic) and paraelectric, KNbO3 is distorted (rhombohedral) and ferroelectric at lower temperatures. LiTaO3 has a ferroelectric transition at lower temperature (950 K) than LiNbO3 (1480 K). More ionic bonding and larger band gap with 5d vs. 4d, due to relativistic effects!")
92
Structures of the Dihydride Dimers M2H4 (M = Mg, Ca, Sr, Ba)
(relative MP2 Energies in kJ/Mol) C2h D2h +19.2 +35.5 0.0 +20.1 +56.0 C2v C3v +115.0 +5.9 0.0 D4h +507.0 +130.4 +86.5 +33.4 C2v J. Am. Chem. Soc. 1993, 115, 11202
 C2h. D2h C2v. C3v D4h C2v. J. Am. Chem. Soc. 1993, 115,")
93
Summary: Understanding Non-VSEPR Structures in d0-Complexes
-d0 complexes with purely -bonded ligands tend to violate VSEPR and related models, in spite of increased ligand repulsion. -while extended VSEPR models are not useful in rationalizing the distorted structures, both MO and VB models are successful for simple homoleptic complexes. --bonding is much more important in d0 complexes than in related p-block complexes, due to the avalability of inner d-orbitals. -the interrelation between -bonding and bond angles is complicated, due to the involvement of different d‑orbitals. -the “anti-Bent’s-rule” structure of TiCl2(CH3)2 is due to Ti‑Cl -bonding! -additional -donor ligands influence the angles between -donor ligands in heteroleptic complexes.
2 is due to Ti‑Cl -bonding! -additional -donor ligands influence the angles between -donor ligands in. heteroleptic complexes.")
94
Thank you for your attention!

95
“Natural Population Analysis” “Natural Bond Orbital Analysis”
Weinhold et al. 1984, 1988 AO basis original basis set occupancy-weighted Löwdin orthogonalization NAO basis “natural atomic orbitals” “natural minimal basis set” (strongly occupied) “natural Rydberg basis set” (sparsely occupied) NHO basis “natural hybrid orbitals” “natural bond orbitals” NBO-Lewis-structure NBO basis “natural resonance theory” (NRT) superposition of NBO-Lewis structures Weinhold et al. 1995 NLMO basis “natürliche lokalisierte Orbitale”
 natural Rydberg basis set (sparsely occupied) NHO basis. natural hybrid orbitals natural bond orbitals NBO-Lewis-structure. NBO basis. natural resonance theory (NRT) superposition of NBO-Lewis structures. Weinhold et al NLMO basis. natürliche lokalisierte Orbitale")
96
NRT Weights (w) for Various Lewis Structures
in Oxo Anions and Sulfur Oxides „wmod“(%) „worig“(%) „walt“(%) „hypervalent“ „original Lewis“ „no-bond/ double-bond“ PO43- 0.0 (0.0) 71.0 (53.1) 20.3 (28.4) SO42- 66.2 (46.9) 23.1 (49.8) ClO4- 60.9 (36.8) 31.4 (60.7) SO3 87.9 (77.3) 5.9 (9.2) SO2 95.2 (90.2) 4.8 (8.6) species NRT at HF/6-31G* (MP2/6-31G*) level Weinhold et al., 1995
 „worig (%) „walt (%) „hypervalent „original. Lewis „no-bond/ double-bond PO (0.0) 71.0 (53.1) 20.3 (28.4) SO (46.9) 23.1 (49.8) ClO (36.8) 31.4 (60.7) SO (77.3) 5.9 (9.2) SO (90.2) 4.8 (8.6) species. NRT at HF/6-31G* (MP2/6-31G*) level. Weinhold et al.,")
97
M C2 C2A C1 H1 H2 H3 M Structure of Mo(butadiene)3
3")
98
Si H + Zr H + Si H . Zr H . Si H - - Zr H
Limits of the Isolobal Principle Si H + Zr H + d(Si-H)=1.488Å, <H-Si-H=120.0º d(Zr-H)=1.800Å, <H-Zr-H=104.8º Si H . Zr H . d(Si-H)=1.500Å, <H-Si-H=110.9º d(Zr-H)=1.862Å, <H-Zr-H=116.6º Si H - - Zr H d(Si-H)=1.563Å, <H-Si-H=93.3º d(Zr-H)=1.902Å, <H-Zr-H=120.0º Review: M. Kaupp Angew. Chemie 2001, 113, 3642.
=1.488Å, <H-Si-H=120.0º. d(Zr-H)=1.800Å, <H-Zr-H=104.8º. Si. H. . Zr. H. . d(Si-H)=1.500Å, <H-Si-H=110.9º. d(Zr-H)=1.862Å, <H-Zr-H=116.6º. Si. H. - - Zr. H. d(Si-H)=1.563Å, <H-Si-H=93.3º. d(Zr-H)=1.902Å, <H-Zr-H=120.0º. Review: M. Kaupp Angew. Chemie 2001, 113,")
99
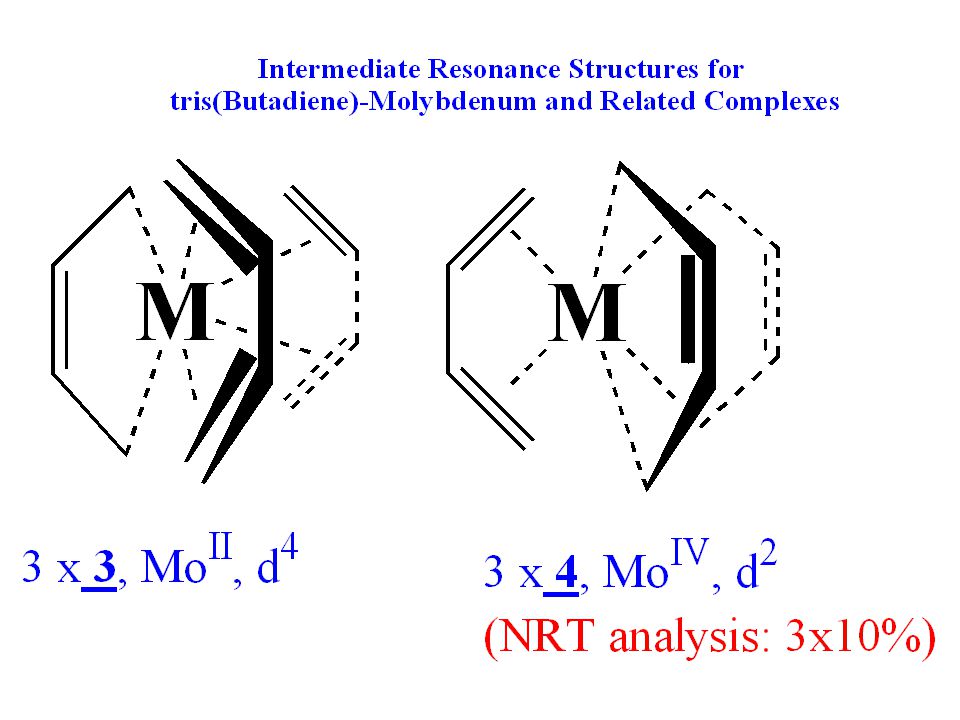
Extreme Resonance Structures for
tris(Butadiene)-Molybdenum and Related Complexes
-Molybdenum and Related Complexes.")
101
a‘‘,e‘ 5p a‘‘ Y4 e‘‘ a‘ 5s 3e‘ 2e‘‘ a‘ 3a‘ Y3 e‘ 2a‘ 4d a‘,e‘,e‘‘ 2e‘ e‘‘ Y2 1a‘‘ a‘‘ 1e‘‘ e‘ 1e‘ Y1 a‘ 1a‘ (C4H6)3 Mo(C4H6)3 C3h Mo
3. Mo(C4H6)3. C3h. Mo.")
102
„Non-Berry“ Rearrangement of TaH5

105
Trends of the s- and d-valence orbitals in the transition metal series
a) radial size from Dirac-Fock calculations (Desclaux)
 radial size. from Dirac-Fock calculations (Desclaux)")
106
Trends of the s- and d-valence orbitals in the transition metal series
b) orbital energies from Dirac-Fock calculations (Desclaux)
 orbital energies. from Dirac-Fock calculations (Desclaux)")
107
Why??

108
Angew. Chem., Int. Ed. Engl. 2001, 40, 3534.

109
On the relative energies of 3d- and 4s orbitals
The 3d orbitals are always lower in energy (and smaller!) than 4s. Stronger electron repulsion in 3d may nevertheless favor 4s occupation.
 than 4s. Stronger electron repulsion in 3d may nevertheless favor 4s occupation.")
110
3p orbital (outermost core shell)
3d orbital (valence shell) Ca atom: radial orbital densities Malrieu et al. Chem. Phys. Lett. 1983, 98, 433.
 Ca atom: radial orbital densities. Malrieu et al. Chem. Phys. Lett. 1983, 98, 433.")
Similar presentations









 Ions: Extended Studies of Hypervalent Species Using the Recoupled Pair Bonding.>")
 Lewis structures and octet rule>")


>")


 Ionic vs. covalent bonding Molecular orbitals and the covalent.>")






