Download presentation
Presentation is loading. Please wait.

1
Finding Host Proteins Required for HIV Replication Abe Brass Partners AIDS Research Center and GI Unit Mass. General Hospital Harvard Medical School

2
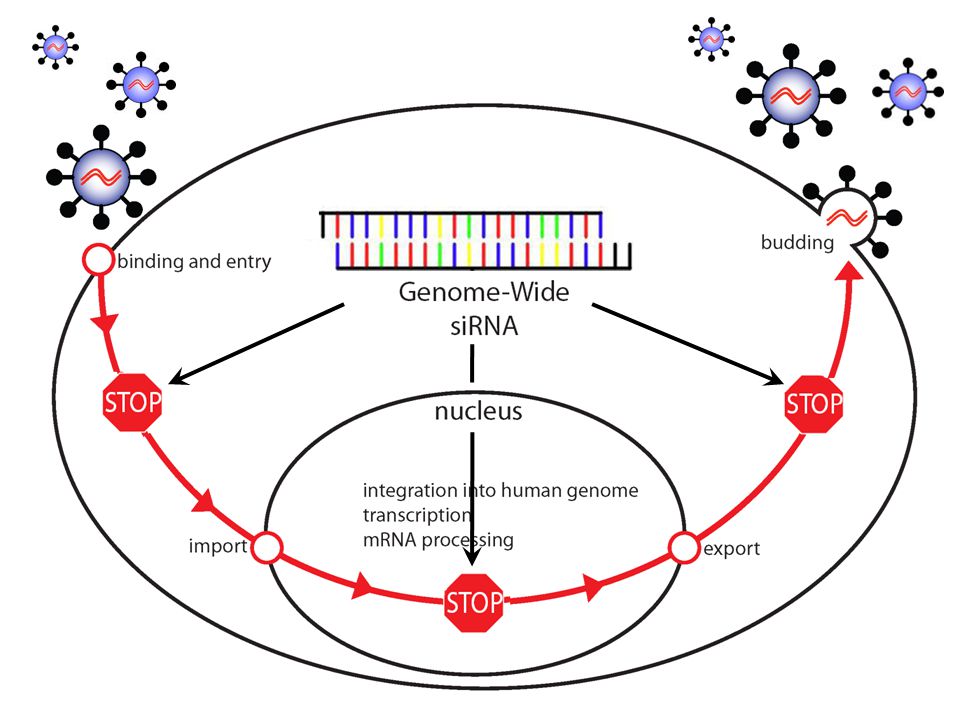
How do we find what HIV needs to replicate?

4
Rationale Employ new methods in mammalian genetics to find host factors that HIV depends upon (HDFs). HDFs provide targets for anti-retroviral therapy and chemical prophylaxis.

5
Rationale HIV may be hard pressed to evade HDF-directed therapies. Comprehensive information about the lifecycle of the virus will benefit the HIV research community.

6
Overview RNAi Mechanism RNAi Tools Finding HIV-dependency factors Three HIV siRNA Screens TNPO3

7
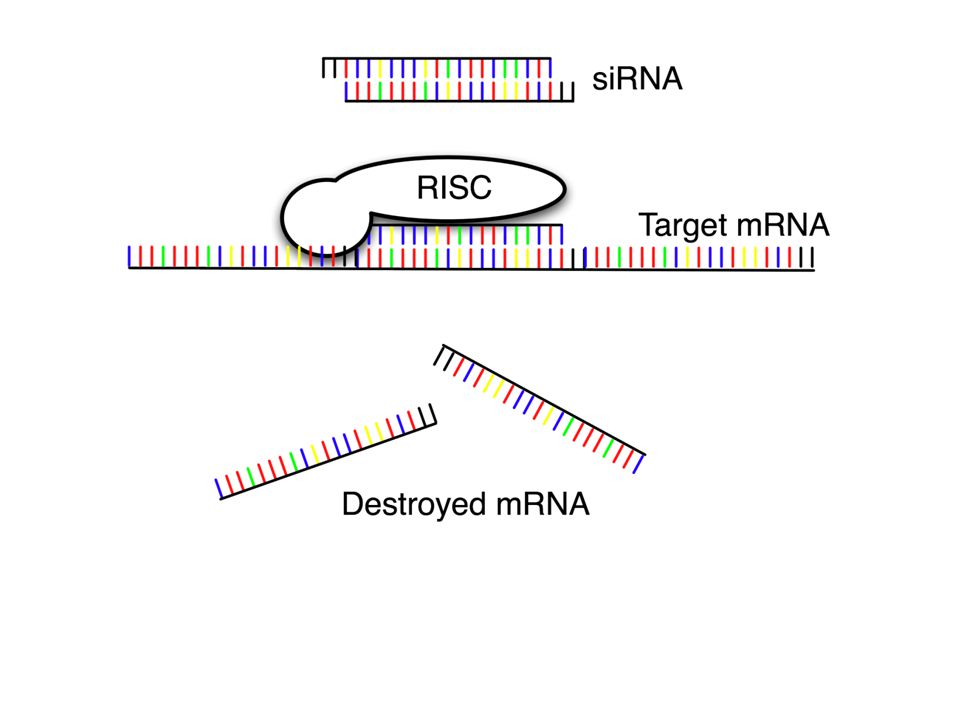
RNAi Mechanism

10
RNAi Tools

11
Genetic Screens Deplete protein expression with shRNAs or siRNAs. Test how depletion impacts phenotype with simple in vitro functional assay. Unbiased whole genome screens bring new targets into the “pipeline”.

12
Genetic Screens The way a genetic screen is designed can profoundly influence which genes are uncovered Different screen platforms yield different results (i.e. libraries, viruses, cell lines, transfection conditions and efficiencies, readouts) Some weak hits may be the most important unlike small molecule screens (knockdown efficiency unknown).
 Some weak hits may be the most important unlike small molecule screens (knockdown efficiency unknown)..")
13
Caveats False positives (OTEs). Present, but minimized through bioinformatic functional clustering, expression studies and reagent redundancy. False negatives. Why didn’t this host factor score? Saturation is the goal, but hard to obtain by generating hypomorphs with the current siRNA technology. Optimized validated reagents will help.

14
shRNA Libraries Whole genome, 3 shRNAs/gene Packaged into Retroviral Pools, Stable knockdown Focused Libraries: Kinases, Ubiquitin pathway shRNAs have unique barcodes Formats : MSCV, lentivirus, Inducible.

15
mir30-5’mir30-3’barcodeLTR mir30-shRNA Retrovirus Integration mir30-shRNA Phenotype Processing of shRNA Target mRNA- single copy knockdown LTR shRNA Libraries

16
Barcode: unique 60 nt sequences that allow the abundance of any particular retroviral shRNA in a complex population to be followed using microarray hybridization. Control Experimental PCR recovery of and color label of barcode Competitive hybridization to barcode microarray shRNA dropped out following selection shRNA enriched following selection

17
siRNA Libraries Arrayed format-”one gene per well” High throughput whole genome screens done with liquid handling robotics. Transiently transfect siRNAs, RISC active for 6 days post transfection. Image based=scanning microscope. Reporter gene=plate reader.

18
Finding HIV-dependency factors

22
CD4 Tat Luciferase Part two Part one

23
siRNA Library Dharmacon: SMARTpool library, 4 siRNAs per pool, whole human genome (21,121 genes). Initial screen done with pools. Validation round done with the four individual siRNAs.

24
Screen Results Out of 21,121 genes, 386 scored 2 SD below control 1.8% hit rate Each of the four individual siRNA were retested 16 genes scored with 4/4 individual siRNAs 44 genes 3/4 99 genes 2/4 115 genes 1/4 273 of 386 siRNA pools were confirmed by at least 1 siRNA (71%) Reagent Redundancy tries to minimize OTEs, but some of the ¼ are “knowns”.
 Reagent Redundancy tries to minimize OTEs, but some of the ¼ are knowns .")
25
Known Host Factors Found in the Screen A4GALT (2/4 siRNAs)DDX3X (2)PSME2 (1) AKT1 (2)ERCC3 (3)PURA (2) AP2M1 (1 )FBXW11 (4)Rab9p40 (3) Arf1 (2)GCN5L2 (1)RANBP1 (1) CD4 (2)H3F3A (1)RelA (4) CD147 (3)HRS (SP)SIP1 (1) CRTC2 (1 )HTATSF1 (1)ST3GAL5 (1) CRTC3 (3 )IKBG (2)TFAP4 (3) CTDP1 (1)La Autoantigen (2)TFE3 (2) CXCR-4 (2)FAPP1 (1)VPRBP (1) CyclinT1 (1) NMT1 (3 ) ZNRD1 (2)
DDX3X (2)PSME2 (1) AKT1 (2)ERCC3 (3)PURA (2) AP2M1 (1 )FBXW11 (4)Rab9p40 (3) Arf1 (2)GCN5L2 (1)RANBP1 (1) CD4 (2)H3F3A (1)RelA (4) CD147 (3)HRS (SP)SIP1 (1) CRTC2 (1 )HTATSF1 (1)ST3GAL5 (1) CRTC3 (3 )IKBG (2)TFAP4 (3) CTDP1 (1)La Autoantigen (2)TFE3 (2) CXCR-4 (2)FAPP1 (1)VPRBP (1) CyclinT1 (1) NMT1 (3 ) ZNRD1 (2)")
26
Biologic Processes

28
Enzymes Found in the Screen ADAM10 (3/4 siRNAs)WNK1 (3)Jak1 (1) DDX55 (3)PSPHL (2)USP26 (2) DDX49 (2)DPM1 (2)OTUD3 (2) ATPV0A1 (2)OST48 (3)LPL (2) GAPVD1(2)PRKX1 (1)HUWE1 (2) PIGH (1)STT3A (2)HERC3 (3) PIGY (2)RNF170 (3)EXOD1(2) ABGL5 (2)FNTA(4)HERC6 (2) FLJ32569 (2)ALKBH8 (2)DDX33 (2) ITPKA (2)NMT1 (3)SET7 (3) MOS (2)DIMT1L(4)ARF1 (2) PIP5K1C (3)C14orf125 (3)CENTG1 (3)
WNK1 (3)Jak1 (1) DDX55 (3)PSPHL (2)USP26 (2) DDX49 (2)DPM1 (2)OTUD3 (2) ATPV0A1 (2)OST48 (3)LPL (2) GAPVD1(2)PRKX1 (1)HUWE1 (2) PIGH (1)STT3A (2)HERC3 (3) PIGY (2)RNF170 (3)EXOD1(2) ABGL5 (2)FNTA(4)HERC6 (2) FLJ32569 (2)ALKBH8 (2)DDX33 (2) ITPKA (2)NMT1 (3)SET7 (3) MOS (2)DIMT1L(4)ARF1 (2) PIP5K1C (3)C14orf125 (3)CENTG1 (3)")
29
Three HIV siRNA Screens

30
CellsKDVirusInfectionReadoutHits Zhou et al HeLa-Bgal24 hHXB248 hr; 96 hr β-gal reporter activation 207 Konig et al 293T48 h NL 4.3 luc vector, VSV-G 24 hrLuc reporter 295 Brass et al TZM-bl, HeLa 72 h HIV-1- IIIB 48 hr; 48 hr in new cells p24 ; reporter activation 280 Comparison of Three HIV siRNA Screens Stephen Goff Cell 135 2008

31
18 13 9 207 280 295 Brass et al Zhou et al Konig et al RelA Med6 Med7 783 total

32
HDFs Found by Two Independent Screens ADRBK1DDX3XMed7 AKT1DMXL1MID1IP1 ANAPC2IDH1 (1)Mre11A ANKRD30A (1)Jak1 (1)Nup153 Cav2MAP4Nup155 CD4Med14Rab28 (1) CHST1Med19RanBP2 CTDP1 (1)Med28RelA CXCR4Med4RNF26 (1) CyclinT1 (1)Med6TCEB3 WNK1TRIM55TNPO3
Mre11A ANKRD30A (1)Jak1 (1)Nup153 Cav2MAP4Nup155 CD4Med14Rab28 (1) CHST1Med19RanBP2 CTDP1 (1)Med28RelA CXCR4Med4RNF26 (1) CyclinT1 (1)Med6TCEB3 WNK1TRIM55TNPO3")
33
Why so little overlap? Different screen platforms yield different results. Technology caveats: False positives (OTEs) and false negatives (reagents not validated). High throughput methodologies Secondary filters of the primary data
 and false negatives (reagents not validated). High throughput methodologies Secondary filters of the primary data.")
34
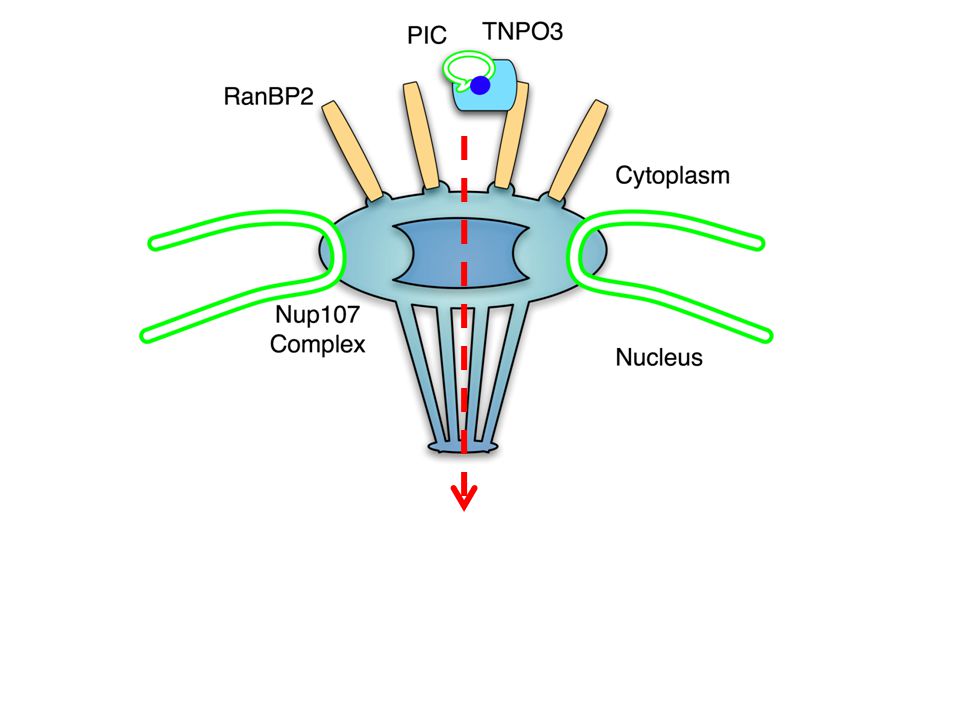
TNPO3

35
SR

36
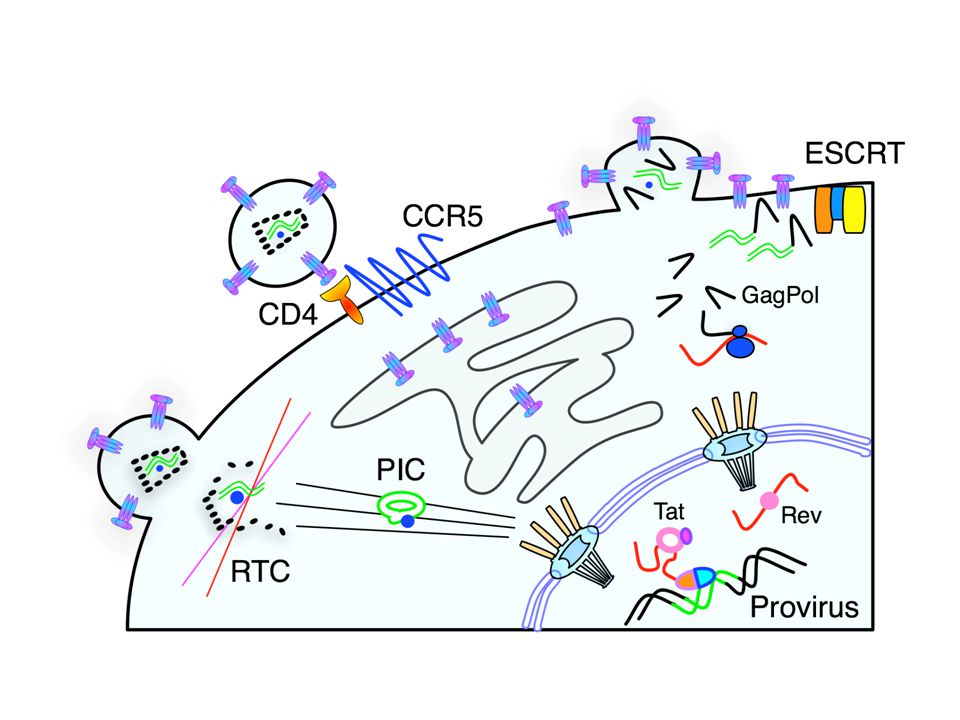
VirionRTCPICProvirusPIC

40
Conclusions Functional enrichment, finding known factors, and confirmation studies suggests the majority of the genes found in the screens impact HIV replication. Clearly conventional validation work is required, but host factor discovery has been accelerated (33+ functionally confirmed host factors in 10 months). Two screens yielded TNPO3, which is very likely the factor that permits HIV-1, HIV-2, EIAV, and SIV access to their host’s genomes.
. Two screens yielded TNPO3, which is very likely the factor that permits HIV-1, HIV-2, EIAV, and SIV access to their host’s genomes..")
41
Thank You Stephen J. Elledge, HHMI, BWH, HMS Judy Lieberman, Nan Yan, Derek Dykxhoorn, IDI, Childrens Hospital Ramnik Xavier, Yair Benita, CCIB, MGH Caroline Shamu and staff, ICCB-L HMSFelipe Diaz-Griffero, DFCI, HMS Bill and Melinda Gates Foundation Harvard CFAR

Similar presentations












>")





 into cells, utilizing.>")






 To discuss the iNUTS and iBOLTS of how mRNAs function in the cytoplasm.>")