Download presentation
Presentation is loading. Please wait.

1
Otto Heinrich Warburg (Friburgo in Brisgovia, 8 ottobre 1883 – Berlino, 1 agosto 1970)
")
2
Warburg O, Posener K, Negelein E. Uber den Stoffwechsel der Tumoren [On metabolism of tumors]. Biochem Z 1924; 152:319–344. Warburg O. The metabolism of tumours. Constable: London; 1930. Warburg O. On the origin of cancer cells. Science 1956; 123:309–314. Warburg O. On respiratory impairment in cancer cells. Science 1956; 124: 269–270.
![Warburg O, Posener K, Negelein E. Uber den Stoffwechsel der Tumoren [On metabolism of tumors].](http://images.slideplayer.com/13/4089612/slides/slide_2.jpg "Biochem Z 1924; 152:319–344. Warburg O. The metabolism of tumours. Constable: London; Warburg O. On the origin of cancer cells. Science 1956; 123:309–314. Warburg O. On respiratory impairment in cancer cells. Science 1956; 124: 269–270..")
5
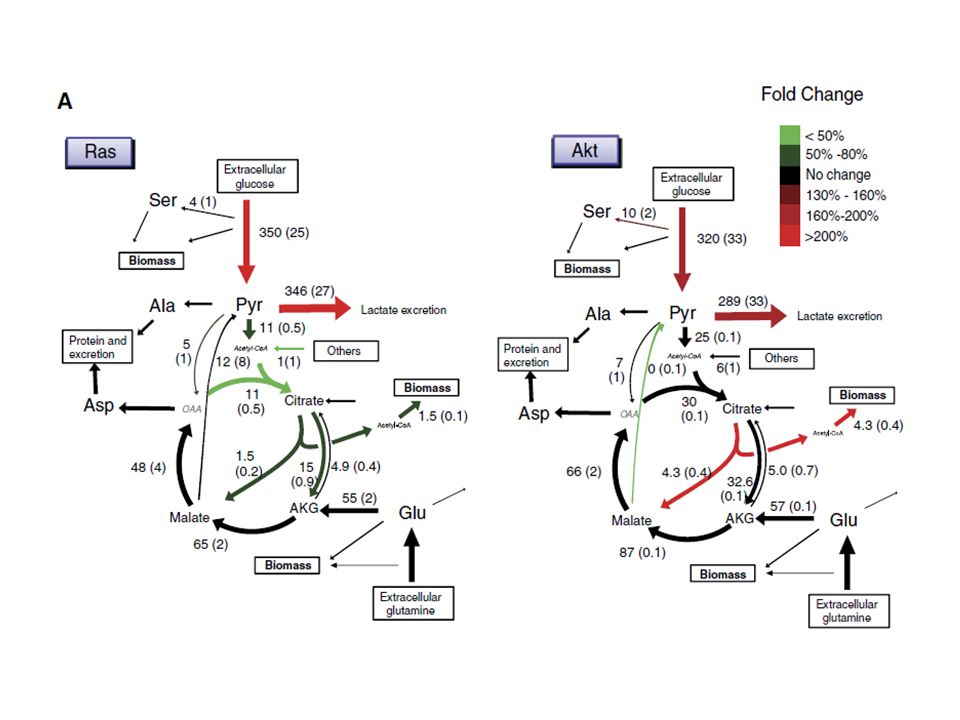
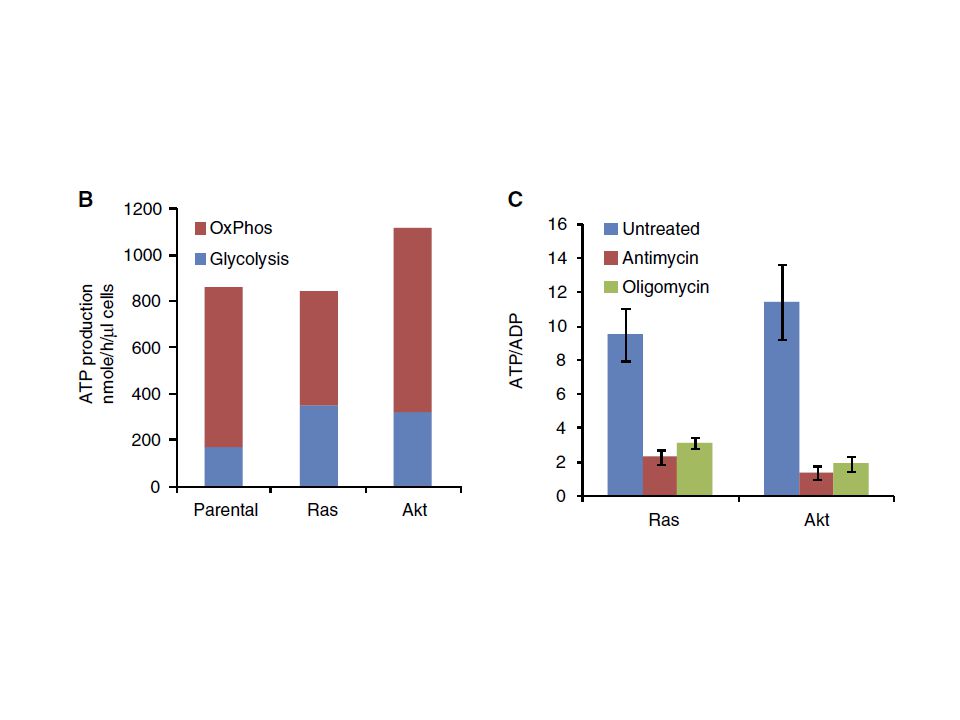
Warburg and co-workers showed in the 1920s that, under aerobic conditions, tumour tissues metabolize approximately tenfold more glucose to lactate in a given time than normal tissues, a phenomenon known as the Warburg effect. However, this increase in aerobic glycolysis in cancer cells is often erroneously thought to occur instead of mitochondrial respiration and has been misinterpreted as evidence for damage to respiration instead of damage to the regulation of glycolysis. In fact, many cancers exhibit the Warburg effect while retaining mitochondrial respiration.

6
90 years after Otto Warburg’s discovery we still ask ourselves “why do cancers have high aerobic glycolysis?” This question, however, can be understood in two different ways: 1. What is the cause of increased aerobic glycolysis in tumor cells? or 2. What is the advantage of increased aerobic glycolysis for tumor cells?

7
Otto Warburg Nobel Prize in Physiology or Medicine 1931. Every year, between 20 and 25 Nobel Laureates spend a week in the Lake Constance area to meet the next generation of leading scientists. The Prime Cause and Prevention of Cancer Lecture at the meeting of the Nobel-Laureates on June 30, 1966 at Lindau, Lake Constance, Germany by Otto Warburg Director, Max Planck-Institute for Cell Physiology, Berlin-Dahlem

9
Warburg effect: reprogramming of gene expression that alters glucose metabolism and enables cells to suppress apoptotic signaling, to grow in hypoxic environments, and to use more glucose for anabolic processes rather than oxidative phosphorylation, even when oxygen is not limiting

10
Cell metabolism Quiescent versus proliferating cells Proliferating versus cancer cells

12
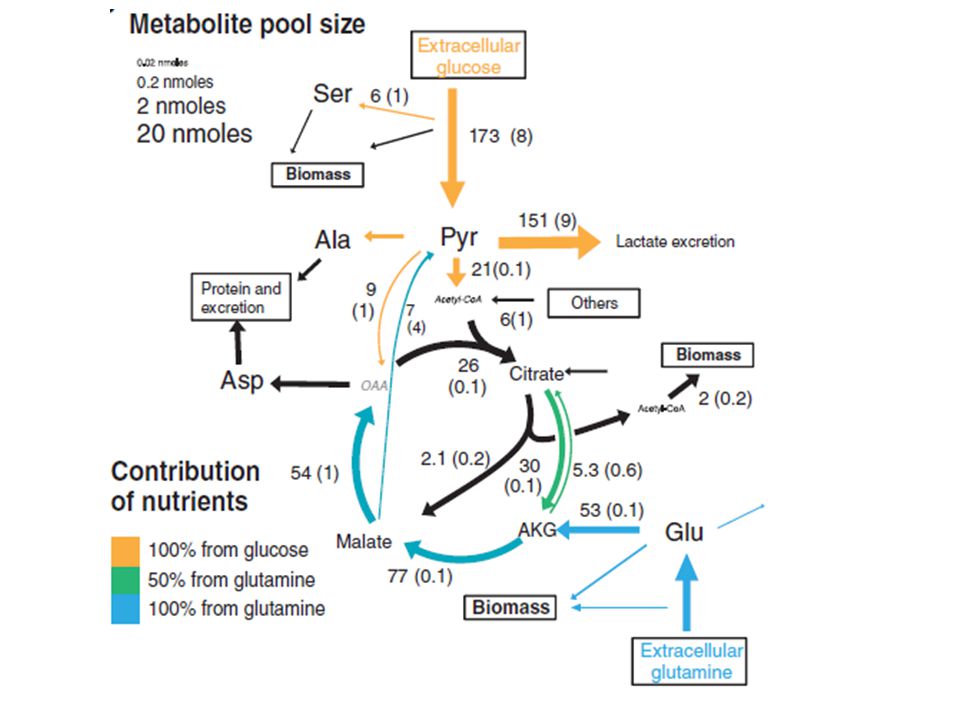
The example illustrates that the biosynthesis of many cellular building blocks requires nutrients in excess of those needed for ATP production alone

14
BIOMASS

16
Glicolisi: Fase Esoergonica Decima reazione: seconda fosforilazione a livello del substrato PEP enolpiruvato (forma enolica) chetopiruvato forma chetonica) Interconversione spontanea Il prodotto finale della 10° reazione è dunque il piruvato in forma chetonica.
 chetopiruvato forma chetonica) Interconversione spontanea Il prodotto finale della 10° reazione è dunque il piruvato in forma chetonica.")
17
Pyruvate kinase isoforms Liver (L) Red blood cell (R) M1, expressed in most adult tissues M2, expressed predominantly in embryonic tissue and tumors PKL gene PKM gene
 Red blood cell (R) M1, expressed in most adult tissues M2, expressed predominantly in embryonic tissue and tumors PKL gene PKM gene")
19
PKM1 and PKM2 differ by only 23 amino acids within a 56-residue alternatively spliced exon (9 or 10, respectively) These amino acids in the M2 isoform constitute an allosteric pocket unique to PKM2 that allows it to bind its activator, fructose 1,6-bisphosphate (FBP), and be regulated by phosphotyrosine-based growth signals
 These amino acids in the M2 isoform constitute an allosteric pocket unique to PKM2 that allows it to bind its activator, fructose 1,6-bisphosphate (FBP), and be regulated by phosphotyrosine-based growth signals")
20
PKM2 exists as either an active tetramer or a relatively inactive dimer or monomer. The latter form is commonly expressed at high levels in tumor cells, and this conformation is favored by binding of phosphotyrosinated proteins, which displace FBP, the glycolytic metabolite that induces formation of the tetrameric form

21
The specific activity of PKM2 that is fully activated by FBP is approximately half that of PKM1. In the absence of FBP, PKM2 had less than one quarter of the activity of PKM1.

22
Most cancer cells studied to date exclusively express PKM2 whereas cells in many normal differentiated tissues express PKM1. This selection for the decreased activity of a rate-limiting glycolytic enzyme appears inconsistent with the increased glucose utilization that is characteristic of cancer cells.

23
This lack of ATP synthesis may allow cells to metabolize glucose by a modified glycolysis that does not generate ATP and provides an advantage to proliferating cells. Historically, efforts to understand aerobic glycolysis stressed the importance of ATP consumption to allow the high rate of glucose metabolism observed in tumor cells. Cells must avoid ATP production in excess of demand to avoid allosteric inhibition of phosphofructokinase and other rate limiting steps in glycolysis that are inhibited by a high ATP/AMP ratio

26
Evidence for an alternative glycolytic pathway in rapidly proliferating cells Matthew G. Vander Heiden 1,2,3,*, Jason W. Locasale 2,3, Kenneth D. Swanson 2, Hadar Sharfi 2, Greg J. Heffron 4, Daniel Amador-Noguez 5, Heather R. Christofk 2, Gerhard Wagner 4, Joshua D. Rabinowitz 5, John M. Asara 2, and Lewis C. Cantley 2,3,† 1 Dana Farber Cancer Institute, Harvard Medical School, Boston, MA 02115 2 Beth Israel Deaconess Medical Center, Division of Signal Transduction and Department of Medicine, Harvard Medical School, Boston, MA 02115 3 Department of Systems Biology, Harvard Medical School, Boston, MA 02115 4 Department of Biological Chemistry and Molecular Pharmacology; Harvard Medical School, Boston, MA 02115 5 Lewis-Sigler Institute for Integrative Genomics and Department of Chemistry, Princeton University, Princeton, NJ 08544 Science. 2010 September 17; 329(5998): 1492–1499.
: 1492–")
27
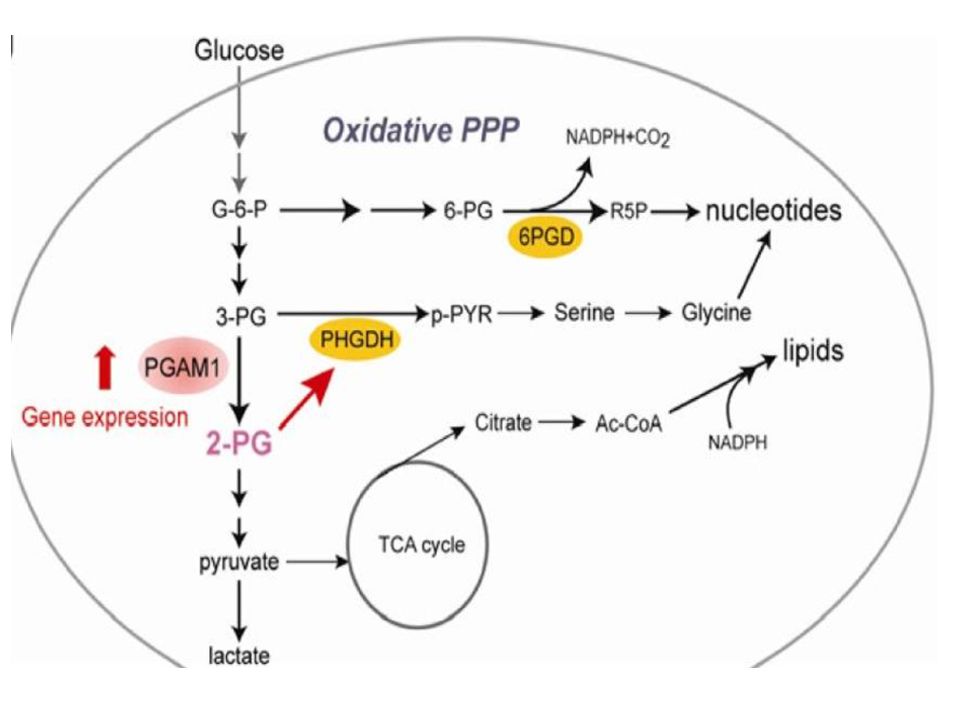
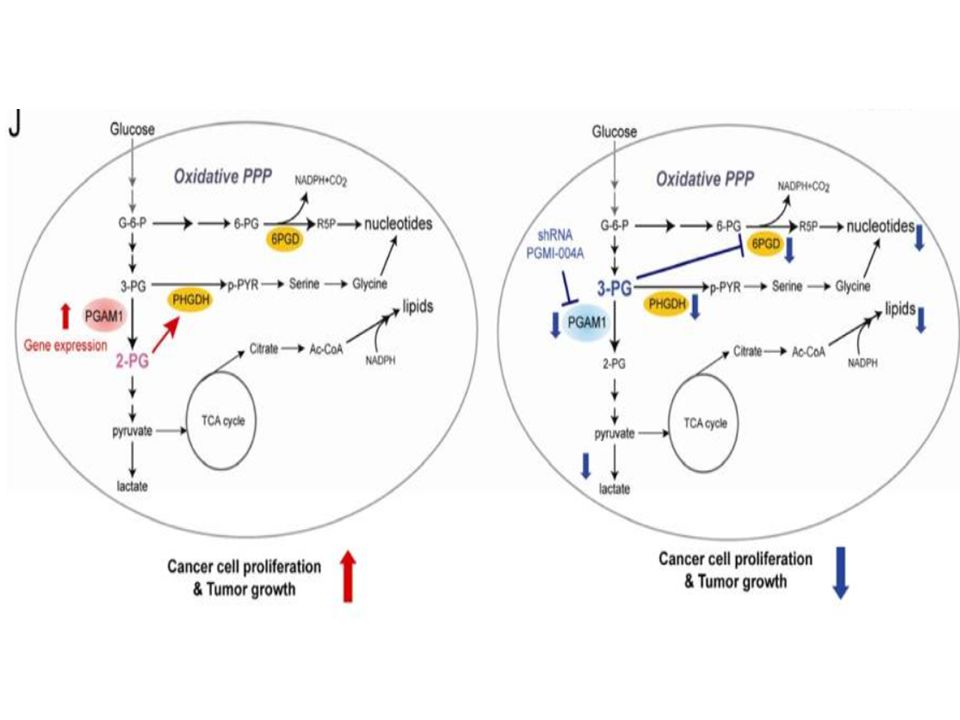
Cancer Cell. 2012 November 13; 22(5): 585–600. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth Taro Hitosugi et al. 3-PG2-PG

31
BIOSINTESI DELLE PIRIMIDINE

33
Ciclo di Krebs: enzimi 1 citrato sintasi 2 aconitasi 3 isocitrato deidrogenasi 4 -chetoglutarato deidrogenasi 5succinil-CoA sintetasi 6succinato deidrogenasi 7fumarasi 8malato deidrogenasi

35
glutaminolysis

36
l’enzima malico catalizza la reazione che concorre a fornire equivalenti riducenti per la sintesi degli acidi grassi

40
AA 07/08 Fase delle decarbossilazioni ossidative (reazioni 3, 4): Reazione n°3: 1 a decarbossilazione ossidativa. La reazione, catalizzata dall’enzima isocitrato deidrogenasi, prevede l’ossidazione dell’isocitrato con formazione di un intermedio, l’ossalsuccinato, dal quale si distacca una molecola di CO 2. Si ha dunque formazione di NAD ridotto e liberazione di una molecola di anidride carbonica. L’ossalsuccinato viene decarbossilato mentre si trova legato all’enzima e non compare mai in forma libera

45
% of total NADH/FADH2 production glutamine60 glucose30 Acetyl-CoA from other sources (fatty acids, amino acids) 10
 10")
46
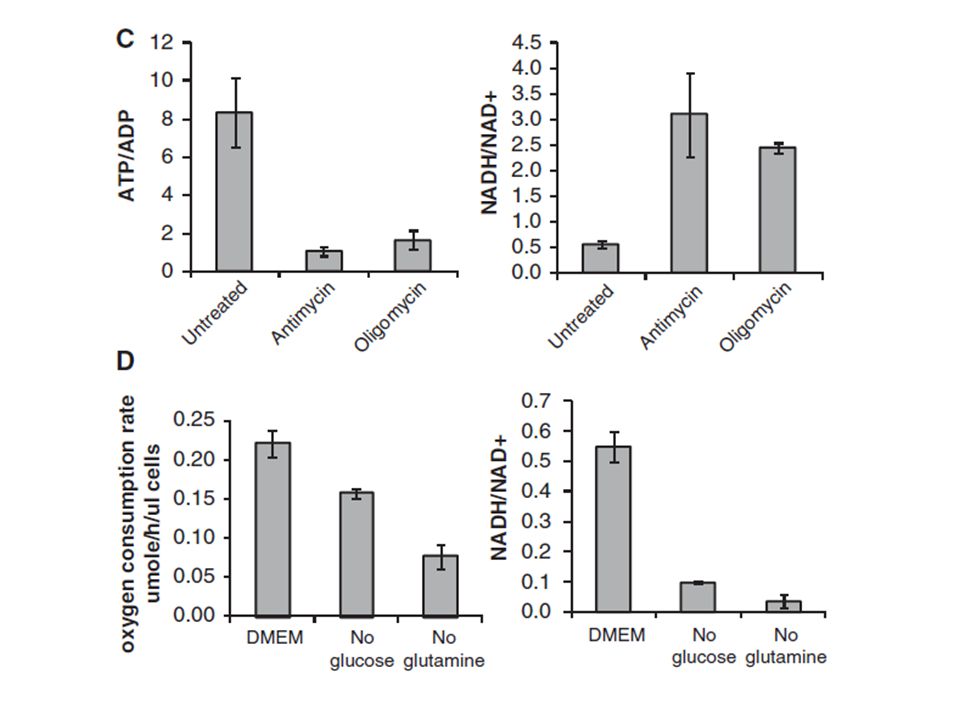
Moreover, we observed that, of total NADH generated in the cytosol, 84% is consumed to reduce pyruvate to lactate. For the remaining 16% of cytosolic high-energy electrons to contribute to ATP production, they must be imported into the mitochondrion via the malate-aspartate shuttle, which produces mitochondrial NADH, or the glycerol-phosphate shuttle, which converts cytosolic NADH into mitochondrial FADH2.

47
Whatever its function, the occurrence of the Warburg effect reflects the activation of oncogenic signaling pathways whose physiological function is to promote glucose uptake and anabolic metabolism.

48
In contrast to glucose, glutamine uptake was not found to be under the direct or indirect control of the PI3K/AKT pathway. Inhibitors of either PI3K or AKT, despite suppressing glucose metabolism in a dose-dependent fashion, had no effect on the glutaminolytic phenotype. In contrast, high level expression of Myc was required to maintain the glutaminolytic phenotype and addiction to glutamine as a bioenergetic substrate. Induction of these key regulatory genes involved in glutaminolysis correlated with the Myc-induced increases in glutamine uptake and glutaminase flux. This increase in glutamine uptake was not a compensatory response to increased glutamine incorporation into proteins as a result of Myc-induced protein synthesis, because most of the additional glutamine carbon taken up after Myc induction was secreted as lactate. Myc-induced reprogramming of intermediate metabolism resulted in glutamine addiction, despite the abundant availability of glucose.

54
Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate Patrick S. Ward 1,2 and Craig B. Thompson 1,* 1 Cancer Biology and Genetics Program, Memorial Sloan-Kettering Cancer Center, New York, NY 10065 2 Cell and Molecular Biology Graduate Group, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA 19104 Cancer Cell. 2012 March 20; 21(3): 297–308.
: 297–308..")
55
glucosio piruvato Lattato o Acetil-CoA PHGDH PSAT PSPH PHGDH: fosfoglicerato deidrogenasi PSAT: fosfoserina aminotransferasi PSPH: fosfoserina fosfoidrolasi GLS-2: glutaminase-2 glutamina Via della serina GLICOLISI GLS-2

57
Nat Genet. 2011 Jul 31;43(9):869-74. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Locasale JW, et al. Department of Systems Biology, Harvard Medical School, Boston, Massachusetts, USA

61
Suppression of PHGDH in MDA-MB-468 cells caused a large reduction in the levels of a-keto-glutarate. In fact, of the major metabolites measured, aKG was the one with the most significant and largest change upon PHGDH suppression, whereas serine levels were not significantly changed. glutamine

64
Science. 2012 May 25;336(6084):1040-4. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK. Broad Institute, Cambridge, MA 02142, USA.

65
We measured the consumption and release (CORE) profiles of 219 metabolites from media across the NCI-60 cancer cell lines, and integrated these data with a preexisting atlas of gene expression. This analysis identified glycine consumption and expression of the mitochondrial glycine biosynthetic pathway as strongly correlated with rates of proliferation across cancer cells. Antagonizing glycine uptake and its mitochondrial biosynthesis preferentially impaired rapidly proliferating cells. Moreover, higher expression of this pathway was associated with greater mortality in breast cancer patients. Increased reliance on glycine may represent a metabolic vulnerability for selectively targeting rapid cancer cell proliferation.

66
Original Article Recurring Mutations Found by Sequencing an Acute Myeloid Leukemia Genome Elaine R. Mardis, Ph.D., Li Ding, Ph.D., David J. Dooling, Ph.D., David E. Larson, Ph.D., Michael D. McLellan, B.S., Ken Chen, Ph.D., Daniel C. Koboldt, M.S., Robert S. Fulton, M.S., Kim D. Delehaunty, B.A., Sean D. McGrath, M.S., Lucinda A. Fulton, M.S., Devin P. Locke, Ph.D., Vincent J. Magrini, Ph.D., Rachel M. Abbott, B.S., Tammi L. Vickery, B.S., Jerry S. Reed, M.S., Jody S. Robinson, M.S., Todd Wylie, B.S., Scott M. Smith, Lynn Carmichael, B.S., James M. Eldred, Christopher C. Harris, B.S., Jason Walker, B.A., B.S., Joshua B. Peck, M.B.A., Feiyu Du, M.S., Adam F. Dukes, B.A., Gabriel E. Sanderson, B.S., Anthony M. Brummett, Eric Clark, Joshua F. McMichael, B.S., Rick J. Meyer, M.S., Jonathan K. Schindler, B.S., B.A., Craig S. Pohl, M.S., John W. Wallis, Ph.D., Xiaoqi Shi, M.S., Ling Lin, M.S., Heather Schmidt, B.S., Yuzhu Tang, M.D., Carrie Haipek, M.S., Madeline E. Wiechert, M.S., Jolynda V. Ivy, M.B.A., Joelle Kalicki, B.S., Glendoria Elliott, Rhonda E. Ries, M.A., Jacqueline E. Payton, M.D., Ph.D., Peter Westervelt, M.D., Ph.D., Michael H. Tomasson, M.D., Mark A. Watson, M.D., Ph.D., Jack Baty, B.A., Sharon Heath, William D. Shannon, Ph.D., Rakesh Nagarajan, M.D., Ph.D., Daniel C. Link, M.D., Matthew J. Walter, M.D., Timothy A. Graubert, M.D., John F. DiPersio, M.D., Ph.D., Richard K. Wilson, Ph.D., and Timothy J. Ley, M.D. Vol 361(11):1058-1066, September 10, 2009
: , September 10,")
67
Study Overview A comparison of the genomic sequence of a tumor sample from a patient with acute myeloid leukemia (AML) and that of a normal skin sample from the same patient revealed an estimated 750 somatic mutations, of which 12 were in the coding sequences of genes and 52 were in conserved regions or regions with regulatory potential Four mutations were found to be recurrent in AML, including mutations in NRAS, NPM1, IDH1, and a conserved region on chromosome 10
 and that of a normal skin sample from the same patient revealed an estimated 750 somatic mutations, of which 12 were in the coding sequences of genes and 52 were in conserved regions or regions with regulatory potential Four mutations were found to be recurrent in AML, including mutations in NRAS, NPM1, IDH1, and a conserved region on chromosome 10")
68
We predicted 1458 tumor-specific point mutations with high confidence; we tested 116 of these with validation sequencing and confirmed 61 of them (53%). Thus, this genome may contain approximately 750 somatic point mutations. We detected mutations in NRAS, NPMc, and IDH1 and a tier 2 mutation on chromosome 10 in more than one AML genome, suggesting that these mutations are not random and are probably important for the pathogenesis of this tumor.

70
The high-throughput sequencing of glioblastoma multiforme tumors identified a novel mutation in IDH1 that was present in 12% of tumors from glioblastoma multiforme patients (Parsons et al., 2008). Further investigation has revealed that this mutation is present in a high proportion of gliomas and secondary glioblastomas, but not in other human malignancies (Balss et al., 2008; Bleeker et al., 2009; Hartmann et al., 2009; Kang et al., 2009; Sanson et al., 2009; Watanabe et al., 2009; Yan et al., 2009). Less common mutations in IDH2 have also been identified in gliomas, and are mutually exclusive with mutations in IDH1 (Hartmann et al., 2009; Sonoda et al., 2009; Yan et al., 2009).
. Less common mutations in IDH2 have also been identified in gliomas, and are mutually exclusive with mutations in IDH1 (Hartmann et al., 2009; Sonoda et al., 2009; Yan et al., 2009)..")
71
Recently, whole genome sequencing of a patient with acute myelogenous leukemia (AML) identified an R132 mutation in IDH1 (Mardis et al., 2009). Sequencing of additional patients revealed that IDH1 is mutated at R132, mainly to histidine and cysteine, in 8% of AML patients, demonstrating that this mutation is not restricted to gliomas, as previously thought (Mardis et al., 2009).
..")
72
AML patients with the IDH1 R132 mutation harbored high serum levels of 2-HG. Maxmen Journal of Experimental Medicine 2010:0:jem.2072iti5-jem.2072iti5 © 2010 Maxmen

74
In cytogenetically normal AML, mutated IDH1 is found in 10.9% of patients, while mutated IDH2 is found in 12.1%. Mutations in IDH1 are almost exclusively found at amino acid arginine 132 and IDH2 mutations are found more frequently at position R140 and less frequently at position R172.

76
TET

78
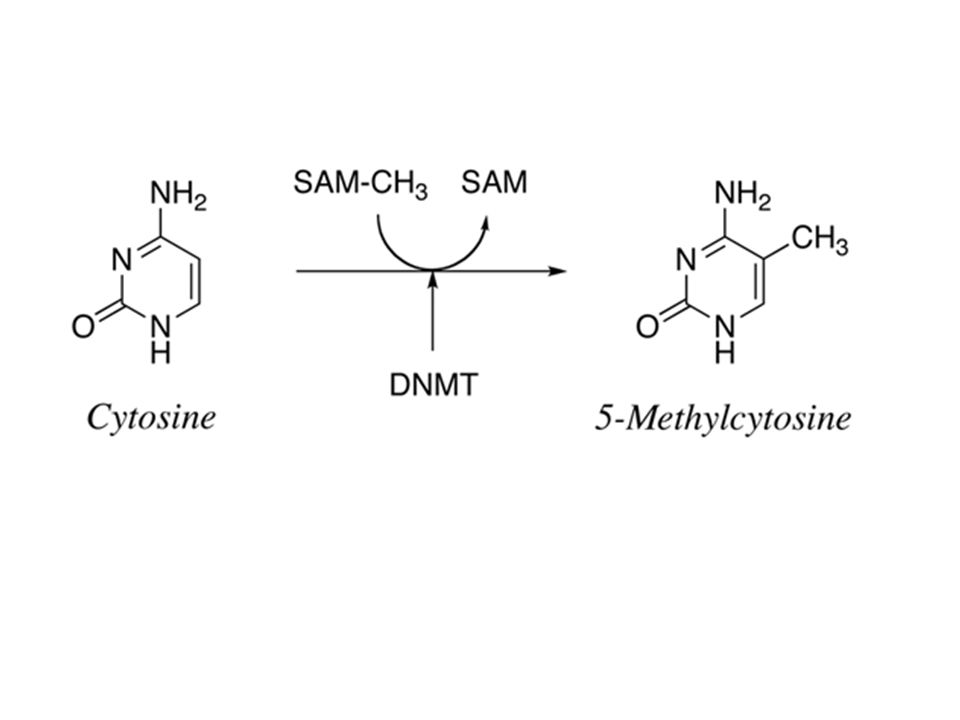
Cancer Cell. 2011 Jan 18;19(1):17-30. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. State Key Laboratory of Genetic Engineering, School of Life Sciences, Shanghai Medical School, Fudan University, Shanghai 20032, China. IDH1 and IDH2 mutations occur frequently in gliomas and acute myeloid leukemia, leading to simultaneous loss and gain of activities in the production of α-ketoglutarate (α-KG) and 2-hydroxyglutarate (2-HG), respectively. Here we demonstrate that 2-HG is a competitive inhibitor of multiple α-KG-dependent dioxygenases, including histone demethylases and the TET family of 5-methlycytosine (5mC) hydroxylases. 2-HG occupies the same space as α-KG does in the active site of histone demethylases. Ectopic expression of tumor-derived IDH1 and IDH2 mutants inhibits histone demethylation and 5mC hydroxylation. In glioma, IDH1 mutations are associated with increased histone methylation and decreased 5-hydroxylmethylcytosine (5hmC). Hence, tumor-derived IDH1 and IDH2 mutations reduce α-KG and accumulate an α-KG antagonist, 2-HG, leading to genome-wide histone and DNA methylation alterations.
 and 2-hydroxyglutarate (2-HG), respectively. Here we demonstrate that 2-HG is a competitive inhibitor of multiple α-KG-dependent dioxygenases, including histone demethylases and the TET family of 5-methlycytosine (5mC) hydroxylases. 2-HG occupies the same space as α-KG does in the active site of histone demethylases. Ectopic expression of tumor-derived IDH1 and IDH2 mutants inhibits histone demethylation and 5mC hydroxylation. In glioma, IDH1 mutations are associated with increased histone methylation and decreased 5-hydroxylmethylcytosine (5hmC). Hence, tumor-derived IDH1 and IDH2 mutations reduce α-KG and accumulate an α-KG antagonist, 2-HG, leading to genome-wide histone and DNA methylation alterations..")
82
A novel inhibitor of mutant IDH1 inhibits human AML cell growth As inhibition of several signalling pathways did not selectively inhibit IDH1mut cells, we performed a computational drug screen using the ZINC library and the published crystal structure of mutant IDH1. By computational screening we identified a potential inhibitor of mutant IDH1 termed 2-[2-[3-(4-fluorophenyl)pyrrolidin-1-yl]ethyl]-1,4- dimethylpiperazine (here termed HMS-101). Computational modeling showed that HMS-101 binds to the isocitrate-binding pocket of mutant IDH1.
pyrrolidin-1-yl]ethyl]-1,4- dimethylpiperazine (here termed HMS-101). Computational modeling showed that HMS-101 binds to the isocitrate-binding pocket of mutant IDH1..")
83
The IC50 for HMS-101 was significantly lower in mouse bone marrow cells transduced with IDH1mut compared to IDH1wt (1 μM vs. 12 μM, respectively, P<.001). Treatment of HoxA9+IDH1mut cells with HMS-101 at 10 μM significantly reduced intracellular R- 2HG levels in vitro. HMS-101 induced apoptosis in IDH1mut cells as evidenced by Annexin V staining and cell cycle analysis by BrDU
. Treatment of HoxA9+IDH1mut cells with HMS-101 at 10 μM significantly reduced intracellular R- 2HG levels in vitro. HMS-101 induced apoptosis in IDH1mut cells as evidenced by Annexin V staining and cell cycle analysis by BrDU.")
Similar presentations














































 October 17, 2003 Haining Zhu Dept. of Molecular.>")




