Download presentation
Presentation is loading. Please wait.

1
First Principle Electronic Structure Calculation Prof. Kim Jai Sam (279-2077) Lab. 공학 4-125 (279-5523) http://ctcp.postech.ac.kr Students : Lee Geun Sik, Yun So Jeong
 Lab. 공학 ( ) Students : Lee Geun Sik, Yun So Jeong.")
4
Optical property of nanocrystal (Yun) optoelectronics, biology Structural phase transition of crystal (Lee) most accurate calculation in phase transition Surface problem (Lee) All require electronic structure calculation of crystal! Our Research Area

7
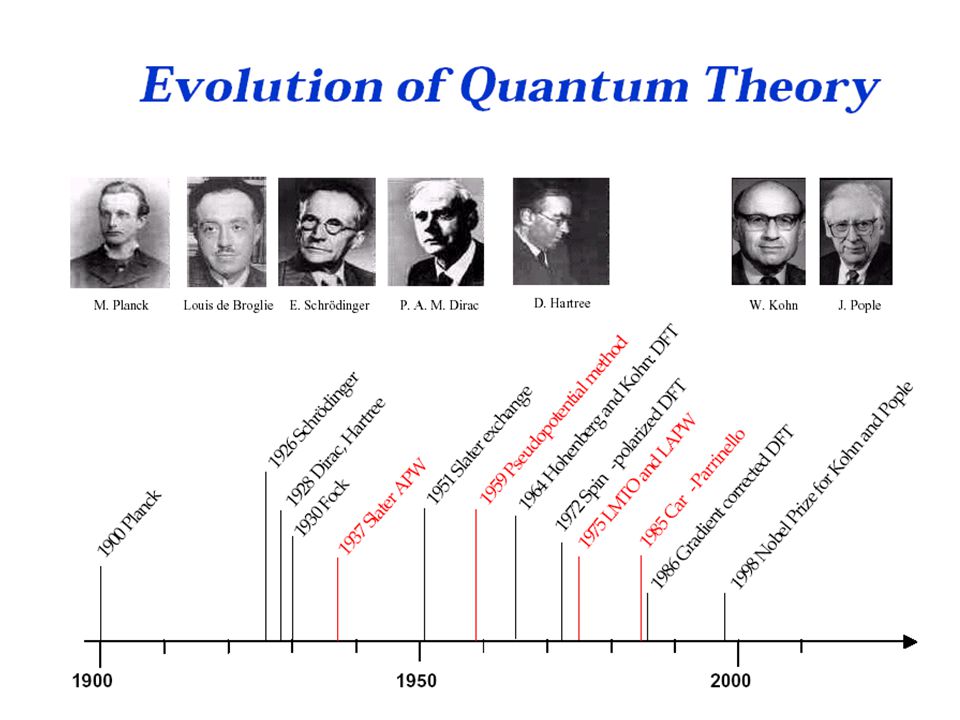
Electronic structure calculation of crystal It was impossible to solve many body problem quantum mechanically. But, with adiabatic approximation (Born-Oppenheimer) and Density Functional Theory (Hohenberg and Kohn 1964, Kohn and Sham 1965), it became possible.
 and Density Functional Theory (Hohenberg and Kohn 1964, Kohn and Sham 1965), it became possible..")
9
Kohn-Sham total-energy functional Kinetic energy of electron Coulomb interaction between ion and electron Coulomb interaction between electrons exchange-correlation energy of electrons static Coulomb interaction between ions DFT says that total energy is a unique functional of the electron density! Minimum energy is the ground state energy!

10
Many electrons problem Self-consistent one-electron equation (Kohn-Sham equation) Variational method
 Variational method")
11
ion Coulomb potential classical electronic Coulomb potential exchange-correlation potential of electron gas (LDA,GGA) Minimize total energy functional self-consistently! Kohn-Sham equation

12
Approximations to the exchange-correlation functional: LDA and GGA

13
Collection of functionals

15
Self-consistent computational procedure

16
Currently using simulation packages in our lab VASP : Pseudopotential, Ultra-soft, PAW, parallel execution in supercomputer. studying CdSe quantum dot system SIESTA : localized orbital basis and pseudopotential, parallel execution, very small basis, handle very large system (nano system). studying now WIEN97 : LAPW method, parallel execution in supercomputer. 9 publications since 2001, mainly TiFe, TiFeH, TiFe(001) system.
. studying now WIEN97 : LAPW method, parallel execution in supercomputer. 9 publications since 2001, mainly TiFe, TiFeH, TiFe(001) system..")
17
Surface electronic structure TiFe (001) Physical Review B, 65, 085410 (2002)
 Physical Review B, 65, (2002)")
18
TiFe (001) Density of States Physical Review B, 65, 085410 (2002)
 Density of States Physical Review B, 65, (2002)")
19
Surface band structure TiFe (001) Physical Review B, 65, 085410 (2002)
 Physical Review B, 65, (2002)")
20
Hydrogen adsorption on TiFe(001) electron density of H/TiFe (001) Int. J. Hydrogen Energy, 27, 403-412 (2002)
.")
21
Angular momentum projected density of states H/TiFe (001) Int. J. Hydrogen Energy, 27, 403-412 (2002)
.")
22
Topology of electronic band I Ag2Se (SG19, P2 1 2 1 2 1 ) CMo2 (SG60, Pbcn) J. Phys. Cond. 15, 2005-2016 (2003)
.")
23
PdSe2 (SG61, Pbca) BFe (SG62, Pnma) J. Phys. Cond. 15, 2005-2016 (2003) Topology of electronic band II
 Topology of electronic band II.")
24
Parallelization of WIEN97 with MPI and SCALAPACK I smaller memory usage with parallel execution!

25
shorter cpu time with parallel execution! Parallelization of WIEN97 with MPI and SCALAPACK II

26
Energy spectrum of nano structure

28
Luminescent Materials I

30
Quantum Dots (optical property) CdSe quantum dot Diameter ~ 4 nm
 CdSe quantum dot Diameter ~ 4 nm")
31
TEM image CdS nanoparticles HRTEM image of single CdS nanoparticle

32
Photoluminescence of bare CdSe and coated CdSe dots Synthetic Metals, 139, 649-652 (2003)
")
33
Applications in biology of optical quantum dots 10 distinguishable colors of ZnS coated CdSe QDs Optical coding and tag based on emission wavelength of ZnS coated CdS QDs

34
Structural phase transition by ab initio method Find the phase which minimize Gibbs free energy, G = E – TS + PV on (P,T) plane. Pressure ↔ volume Temperature ↔ entropy of phonon, harmonic approximation Helmholtz free energy requires phonon density of states, g(ω).
..")
35
Phonon band structure and density of states Solid curve: theoretical calculation Open circle: experimental result J. Chem. Phys. 118, 10174 (2003) MgO
 MgO.")
36
Pressure and Temperature phase diagram B1:NaCl structure B2:CsCl structure Theoretical results agree with experiments quite well! MgO J. Chem. Phys. 118, 10174 (2003)
.")
37
Future Plan Quantum computing quantum dot is one of candidates for qubit. optical properties of quantum dot TDDFT (Time Dependent DFT) calculate electronic structure for excited states. Surface physics : catalysis, hydrogen storage
 calculate electronic structure for excited states. Surface physics : catalysis, hydrogen storage.")
Similar presentations











 www2.le.ac.uk/departments/physics/people/mervynroy.>")
. Chapter.>")



! Hψ = Eψ A differential (operator) eigenvalue equation H.>")





>")



