Download presentation
Presentation is loading. Please wait.

1
Challenges in computational phylogenetics Tandy Warnow Radcliffe Institute for Advanced Study University of Texas at Austin

2
Phylogeny From the Tree of the Life Website, University of Arizona Orangutan GorillaChimpanzee Human

3
Ringe-Warnow Phylogenetic Tree of Indo-European

4
Major methods for phylogeny reconstruction Biology: Polynomial time methods (good enough for small datasets), and local search heuristics for NP-hard optimization problems Linguistics: exact algorithms for NP-hard optimization problems
, and local search heuristics for NP-hard optimization problems Linguistics: exact algorithms for NP-hard optimization problems")
5
Evolution informs about everything in biology Big genome sequencing projects just produce data -- so what? Evolutionary history relates all organisms and genes, and helps us understand and predict –interactions between genes (genetic networks) –drug design –predicting functions of genes –influenza vaccine development –origins and spread of disease –origins and migrations of humans
 –drug design –predicting functions of genes –influenza vaccine development –origins and spread of disease –origins and migrations of humans.")
6
DNA Sequence Evolution AAGACTT TGGACTTAAGGCCT -3 mil yrs -2 mil yrs -1 mil yrs today AGGGCATTAGCCCTAGCACTT AAGGCCTTGGACTT TAGCCCATAGACTTAGCGCTTAGCACAAAGGGCAT TAGCCCTAGCACTT AAGACTT TGGACTTAAGGCCT AGGGCATTAGCCCTAGCACTT AAGGCCTTGGACTT AGCGCTTAGCACAATAGACTTTAGCCCAAGGGCAT

7
Molecular Systematics TAGCCCATAGACTTTGCACAATGCGCTTAGGGCAT UVWXY U VW X Y

8
Basic challenges in molecular phylogenetics Most favored approaches attempt to solve hard optimization problems such as maximum parsimony and maximum likelihood - can we design better methods? DNA sequence evolution may be too “noisy” - perhaps we need new types of data? Many equally good solutions for a given dataset - how can we figure out “truth”? Not all evolution is tree-like - how can we detect and infer reticulate evolution?

9
Main research foci Solving maximum parsimony and maximum likelihood more effectively “Fast converging methods” Gene order and content phylogeny Reticulate evolution Visualizing large phylogenies Data mining on sets of trees

10
Gene Order/Content Phylogeny Group leader: Bernard Moret Software: (1) simulating genome evolution on trees (2) GRAPPA: Genome Rearrangement Analysis using Parsimony and other Phylogenetic Algorithms Currently limited to equal content genomes Ongoing research: handling unequal gene content
 simulating genome evolution on trees (2) GRAPPA: Genome Rearrangement Analysis using Parsimony and other Phylogenetic Algorithms Currently limited to equal content genomes Ongoing research: handling unequal gene content")
11
Reticulate Evolution

12
Some of our projects Divide-and-conquer strategies for maximum parsimony and maximum likelihood Using “rare genomic changes” for deep evolution Consensus/clustering methods for sets of optimal trees Detection and reconstruction of reticulate evolution (All projects are joint with biologists and computer scientists at various universities, and are part of the new ITR grant)
")
13
Coping with NP-hard problems Since NP-hard problems may not be solvable in polynomial time, the options are: –Solve the problem exactly (but use lots of time on some inputs) –Use heuristics which may not solve the problem exactly (and which might be computationally expensive, anyway)
 –Use heuristics which may not solve the problem exactly (and which might be computationally expensive, anyway)")
14
General comments for NP-hard optimization problems Getting exact solutions may not be possible for some problems on some inputs, without spending a great deal of time. You may not know when you have an optimal solution, if you use a heuristic. Sometimes exact solutions may not be necessary, and approximate solutions may suffice. (But this may not be true for biology.)
.")
15
DNA Sequence Evolution AAGACTT TGGACTTAAGGCCT -3 mil yrs -2 mil yrs -1 mil yrs today AGGGCATTAGCCCTAGCACTT AAGGCCTTGGACTT TAGCCCATAGACTTAGCGCTTAGCACAAAGGGCAT TAGCCCTAGCACTT AAGACTT TGGACTTAAGGCCT AGGGCATTAGCCCTAGCACTT AAGGCCTTGGACTT AGCGCTTAGCACAATAGACTTTAGCCCAAGGGCAT

16
Major phylogeny reconstruction methods In biology: mostly hill-climbing heuristics that attempt to solve NP-hard optimization problems (maximum parsimony or maximum likelihood) In historical linguistics: much less is established, but an exact solution to an NP-hard problem looks very promising.
 In historical linguistics: much less is established, but an exact solution to an NP-hard problem looks very promising.")
17
Maximum Parsimony ACT GTTACA GTA ACA ACT GTA GTT ACT ACA GTT GTA

18
Maximum Parsimony ACT GTT GTA ACA GTA 1 2 2 MP score = 5 ACA ACT GTA GTT ACAACT 3 1 3 MP score = 7 ACT ACA GTT GTA ACAGTA 1 2 1 MP score = 4 Optimal MP tree

19
Maximum Parsimony: computational complexity ACT ACA GTT GTA ACAGTA 1 2 1 MP score = 4 Finding the optimal MP tree is NP-hard Optimal labeling can be computed in linear time O(nk)
")
20
Maximum Parsimony Given a set S of strings of the same length over a fixed alphabet, find a tree T leaf-labelled by S and with all internal nodes labelled by strings of the same length over the same alphabet which minimizes the sum of the edge lengths. Motivation: seeks to minimize the total number of point mutations needed to explain the data NP-hard

21
Solving MP (maximum parsimony) and ML (maximum likelihood) Phylogenetic trees MP score Global optimum Local optimum Why are MP and ML hard? The search space is huge -- there are (2n-5)!! trees, it is easy to get stuck in local optima, and there can be many optimal trees. Why try to solve MP or ML? Our experimental studies show that polynomial time algorithms don’t do as well as MP or ML when trees are big and have high rates of evolution. Why solve MP and ML well? Because trees can change in biologically significant ways with small changes in objective criterion. (Open problem!)
!. trees, it is easy to get stuck in local optima, and there can be many optimal trees. Why try to solve MP or ML. Our experimental studies show that polynomial time algorithms don’t do as well as MP or ML when trees are big and have high rates of evolution. Why solve MP and ML well. Because trees can change in biologically significant ways with small changes in objective criterion. (Open problem!).")
22
MP/ML heuristics Time MP score of best trees Performance of hill-climbing heuristic Fake study

23
Speeding up MP/ML heuristics Time MP score of best trees Performance of hill-climbing heuristic Desired Performance Fake study

24
Using divide-and-conquer for MP and ML Conjecture: better (more accurate) solutions will be found in less time, if we analyze a small number of smaller subsets and then combine solutions Need: –1. techniques for decomposing datasets, –2. base methods for subproblems, and –3. techniques for combining subtrees

25
Comparison between TBR and the Ratchet Quite dramatic differences -- the Ratchet finds better trees than the best ways of running TBR branch-swapping, on all our datasets Even the Ratchet can take too long on some datasets! Ochoterena dataset: 834 DNA sequences

26
The DCM3 technique for speeding up MP/ML searches

27
Strict Consensus Merger (SCM)
")
28
DCM3-boosting a base method 1.Decompose the dataset into smaller, overlapping subsets, using DCM3 2.Construct phylogenetic trees on the subsets using a base method 3.Merge the subtrees into a single tree using the Strict Consensus Merger 4.Use PAUP* constrained search to refine the resultant tree

29
What we found I-DCM3-TBR is much faster than TBR on all the datasets we examined I-DCM3-Ratchet is better than the Ratchet, but by less (depends on dataset) I-DCM3-ML improves upon ML using PAUP* ML searches (by a huge amount)
 I-DCM3-ML improves upon ML using PAUP* ML searches (by a huge amount)")
30
What we found DCM3-TBR is much faster than TBR on all the datasets we examined DCM3-Ratchet is better than the Ratchet, but by less (depends on dataset) DCM3-ML improves upon ML using PAUP* ML searches (by a huge amount)
 DCM3-ML improves upon ML using PAUP* ML searches (by a huge amount)")
31
New technique: Iterative DCM3 Repeat: 1. Apply base method for a specified number of iterations. 2. Obtain a DCM3-decomposition based upon the current best tree (the “guide tree” ). 3. Apply base method to subproblems, and merge subtrees using the strict consensus merger. 4. Refine the tree. Variants we have examined: I-DCM3(TBR) and I-DCM3(Ratchet).
. 3. Apply base method to subproblems, and merge subtrees using the strict consensus merger. 4. Refine the tree. Variants we have examined: I-DCM3(TBR) and I-DCM3(Ratchet)..")
32
Popular heuristics PAUP*4.0 hill-climbing heuristics: –Phase 1: do greedy insertions, with limited TBR, to get good starting trees –Phase 2: do TBR branch swapping on the best trees obtained in phase I. Ratchet: –Do standard TBR hillclimbing until stuck in local optima. –Then reweight characters and do TBR hill-climbing to get out of local optima. –Go back to original character set, and repeat.

33
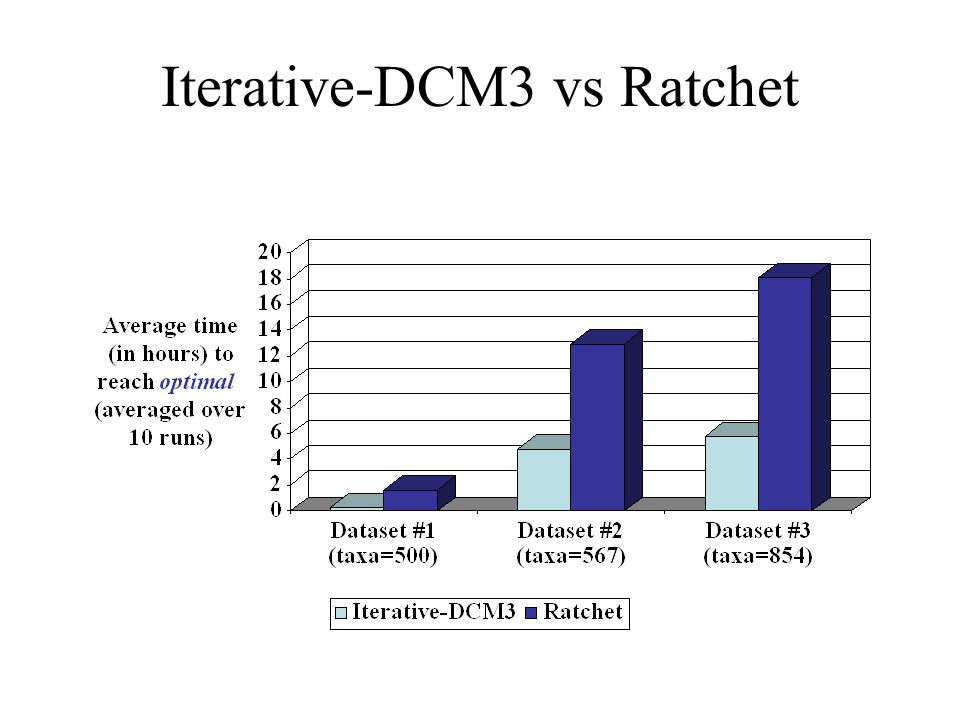
rbcL500 dataset: 500 DNA sequences All 10 runs of Iterative-DCM3 find trees with current best score within75 minutes, whereas Ratchet takes at least 3 hours

34
Gutell dataset: 854 rRNA sequences Iterative-DCM3 trials find trees of MP score 103210 in 30 hours, whereas ratchet500 trials take 45 hours to find trees of same score

35
Iterative-DCM3 vs Ratchet

37
Conclusions I-DCM3 finds trees with MP scores at least as good as Ratchet at every point in time (within first few hours, I-DCM3 is always better) On all datasets I-DCM3 finds good MP trees very quickly Improvements over TBR-based analyses even better
 On all datasets I-DCM3 finds good MP trees very quickly Improvements over TBR-based analyses even better")
38
Ringe-Warnow Phylogenetic Tree of Indo-European

39
Historical Linguistic Data A character is a function that maps a set of languages, L, to a set of states. Three kinds of characters: –Phonological (sound changes) –Lexical (meanings based on a wordlist) –Morphological (grammatical features)
 –Lexical (meanings based on a wordlist) –Morphological (grammatical features).")
40
Cognate Classes Two words w 1 and w 2 are in the same cognate class, if they evolved from the same word through sound changes. French “champ” and Italian “champo” are both descendants of Latin “campus”; thus the two words belong to the same cognate class. Spanish “mucho” and English “much” are not in the same cognate class.

41
Phylogenies of Languages Languages evolve over time, just as biological species do (geographic and other separations induce changes that over time make different dialects incomprehensible -- and new languages appear) The result can be modelled as a rooted tree The interesting thing is that many characteristics of languages evolve without back mutation or parallel evolution -- so a “perfect phylogeny” is possible!
 The result can be modelled as a rooted tree The interesting thing is that many characteristics of languages evolve without back mutation or parallel evolution -- so a perfect phylogeny is possible!")
42
Perfect Phylogeny A phylogeny T for a set S of taxa is a perfect phylogeny if each state of each character occupies a subtree (no character has back-mutations or parallel evolution)
")
43
“Homoplasy-Free” Evolution (perfect phylogenies) YES NO
 YES NO")
44
The Perfect Phylogeny Problem Given a set S of taxa (species, languages, etc.) determine if a perfect phylogeny T exists for S. The problem of determining whether a perfect phylogeny exists is NP-hard (McMorris et al. 1994, Steel 1991).
..")
45
The Indo-European (IE) Dataset 24 languages 22 phonological characters, 15 morphological characters, and 333 lexical characters Total number of working characters is 390 (multiple character coding, and parallel development) A phylogenetic tree T on the IE dataset (Ringe, Taylor and Warnow) T is compatible with all but 22 characters: 16 (18) monomorphic and 6 polymorphic Resolves most of the significant controversies in Indo-European evolution; shows however that Germanic is a problem (not treelike)
 Dataset 24 languages 22 phonological characters, 15 morphological characters, and 333 lexical characters Total number of working characters is 390 (multiple character coding, and parallel development) A phylogenetic tree T on the IE dataset (Ringe, Taylor and Warnow) T is compatible with all but 22 characters: 16 (18) monomorphic and 6 polymorphic Resolves most of the significant controversies in Indo-European evolution; shows however that Germanic is a problem (not treelike)")
46
Improving the model Detected borrowing is not a problem, but undetected borrowing is. We need to work with “networks” rather than trees!

47
Phylogenetic Networks

48
Networks and Trees

49
Character Compatibility on Networks Let N=(V,E) be a phylogenetic network on L, T(N) the set of trees induced by N, and let c:L ! Z be a character. Then c is said to be compatible on N if c is compatible on at least one of the trees in T(N). We can test the compatibility of a character c on a tree T with n leaves in O(n) time, and hence in O(n3 B ) on a network with B non-tree edges
. We can test the compatibility of a character c on a tree T with n leaves in O(n) time, and hence in O(n3 B ) on a network with B non-tree edges.")
50
Perfect Phylogenetic Networks (PPN) All characters are compatible on the network
 All characters are compatible on the network")
51
The Main Problem Given a set L of languages, and a set C of characters, construct a minimum size PPN for L. Our approach: –Testing whether a network is perfect. –Adding edges to a given tree to form a PPN, as a heuristic for solving the main problem.

52
Minimum Size PPN (MSPPN) Input: A set L of n languages, set C of k r-state characters defined on L, and bound B 2 Z. Question: Does there exist a PPN N on L, such that N contains at most B non-tree edges?

53
The MSPPN Problem When B=0 the problem is the perfect phylogeny problem, and hence: –NP-hard in general – solvable in polynomial time for fixed r or fixed k

54
The Indo-European (IE) Dataset 24 languages 22 phonological characters, 15 morphological characters, and 333 lexical characters Total number of working characters is 390 (multiple character coding, and parallel development) A phylogenetic tree T on the IE dataset (Ringe, Taylor and Warnow) T is compatible with all but 22 characters: 16 (18) monomorphic and 6 polymorphic
 Dataset 24 languages 22 phonological characters, 15 morphological characters, and 333 lexical characters Total number of working characters is 390 (multiple character coding, and parallel development) A phylogenetic tree T on the IE dataset (Ringe, Taylor and Warnow) T is compatible with all but 22 characters: 16 (18) monomorphic and 6 polymorphic")
55
Phylogenetic Analysis of the IE Dataset Preprocessing of the set of characters: 16 incompatible characters were found Pruning the tree: the resulting tree had 9 leaves and 16 edges Finding candidate non-tree edges: the set contained 72 possible edges Solving the MIPPN problem on the resulting tree and set of characters: the program computed the 15 possible solutions in about 8 hours

56
Phylogenetic Network of the IE Dataset

57
Open problems Minimum size PPN: NP-hard in general, but polynomial for some fixed-parameter variants? Minimum increment to a PPN: what computational complexity?

58
Major challenges remaining Detecting reticulate evolution in biology, and reconstructing accurate phylogenies in the presence of reticulation Getting sufficiently accurate trees in reasonable amounts of time, for large datasets. Analyzing new types of data (e.g., gene order and content).
..")
59
Acknowledgements Funding: NSF, the David and Lucile Packard Foundation, and the Radcliffe Institute for Advanced Study Collaborators: Bernard Moret and Tiffani Williams (UNM CS), Donald Ringe (Penn Linguistics) Students: Usman Roshan and Luay Nakhleh (UT-Austin)
, Donald Ringe (Penn Linguistics) Students: Usman Roshan and Luay Nakhleh (UT-Austin)")
60
Phylolab, U. Texas Please visit us at http://www.cs.utexas.edu/users/phylo/

Similar presentations






 of Life: Procedures, Problems, and Prospects Presented by Usman Roshan.>")












