Download presentation
Presentation is loading. Please wait.

1
Myelodysplastic and Myelodysplastic/ Myeloproliferative Neoplasms
Myeloproliferative diseases, in general, are disorders in which proliferation of hematopoietic cells outpaces apoptosis, and cellular elements in the blood are increased while the morphology of hematopoiesis is near normal. Myelodysplastic diseases or syndromes are disorders in which apoptosis predominates, hematopoiesis is ineffective, and cytopenias occur. The myelodysplastic/myeloproliferative disorders show features of both, with variable increases in cells, as well as cytopenias and morphologic dysplasia.

2
Types of Abnormal Cellular Maturation
Dyserythropoiesis : Nuclear fragmentation or karyorrhexis, multinuclearity, nuclear budding or bridging,basophilic stippling, and ring sideroblasts. Erythrocytic abnormalities in the blood film include presence of oval macrocytes, anisochromia, basophilic stippling, dacryocytes, and reticulocytopenia.

5
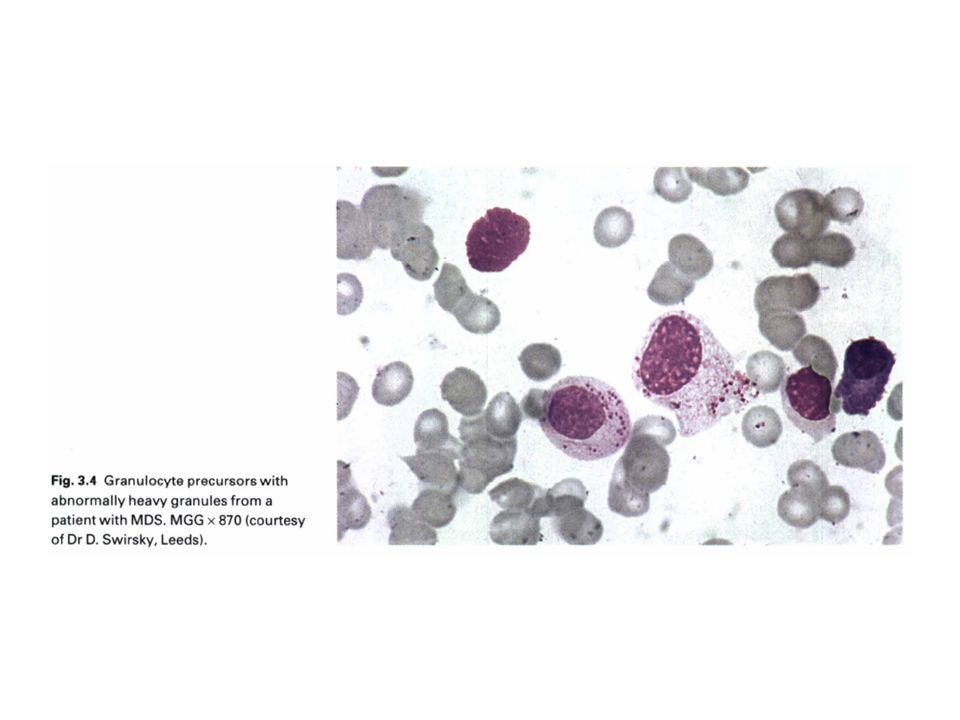
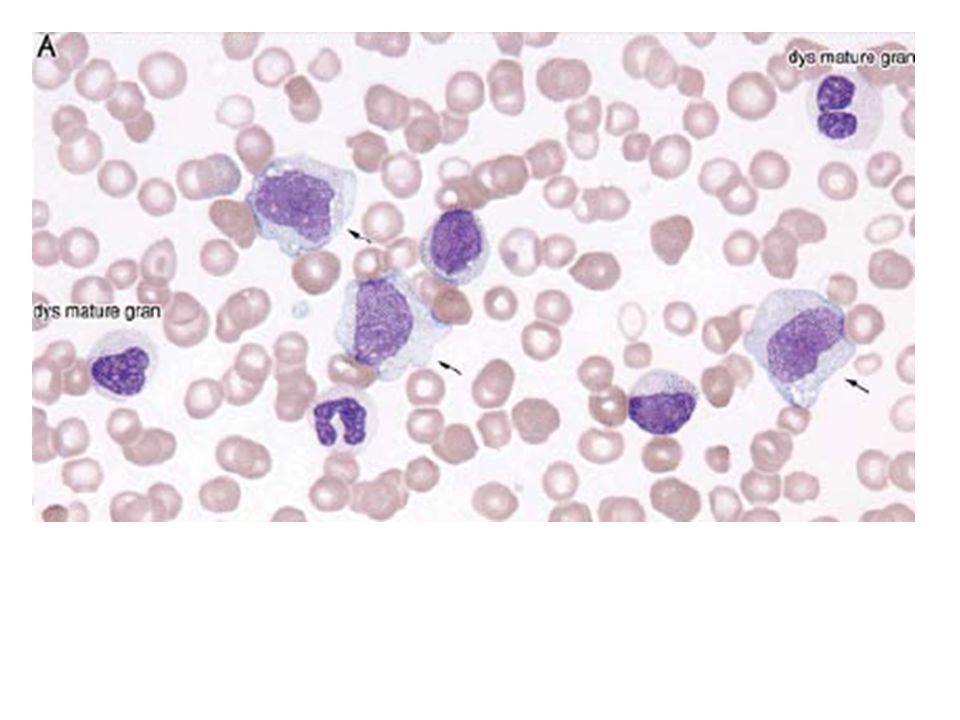
Dysgranulopoiesis: hypogranulation,nuclear hyposegmentation with increased chromatin condensation, occasionally abnormal large azurophilic granules. Dysmegakaryocytopoiesis: large megakaryocytes with unsegmented nuclei, micromegakaryocytes, and megakaryocytes with two or more small, unconnected nuclei .Megakaryocytes may be decreased in number. In the blood film, giant hypogranular platelets are frequent, and micromegakaryocytes are seen rarely.

8
Pseudo–Pelger-Huët neutrophil

10
A dysplastic megakaryocyte showing unconnected nuclear lobes

11
Myelodysplastic/Myeloproliferative Neoplasms
Chronic Myelomonocytic Leukemia Atypical Chronic Myeloid Leukemia, BCR-ABL1 Negative Juvenile Myelomonocytic Leukemia Myelodysplastic/Myeloproliferative Disease, Unclassifiable

13
Chronic Myelomonocytic Leukemia
CMML is a clonal stem cell disorder in which the predominant feature is persistent monocytosis (>1 × 109/L for longer than 3 months) for which other causes have been excluded. Blast percentages typically are <5% in the blood and <10% in the marrow, and such cases are referred to as CMML-1. The subcategory CMML-2 refers to cases with 5%–19% blasts in the blood, with 10%–19% in the marrow, or with the presence of Auer rods.
 for which other causes have been excluded. Blast percentages typically are <5% in the blood and <10% in the marrow, and such cases are referred to as CMML-1. The subcategory CMML-2 refers to cases with 5%–19% blasts in the blood, with 10%–19% in the marrow, or with the presence of Auer rods.")
14
CMML درCMML تقریبا همیشه بیشتراز 10% مونوسیت وجود دارد اما در CML به ندرت از 2-3% تجاوز می کند. درCMML کروموزوم فیلادلفیا وجود ندارد و دیس پلازی یک یاچند رده میلوئیدی وجود دارد و کمتر از 20درصد سلولهای BM را بلاست و پرومونوسیتها تشکیل می دهد. هرگاه تعداد ائوزینوفیلها از /μlبیشتر گردد WHO زیرگروه CMML همراه با ائوزینوفیلی پیشنهاد می کند. از نظر ایمونوفنوتیپ:CD13,CD33مثبت وCD14,CD64,CD68به صورت متغییر مثبت است. ناهنجاریهای سیتوژنتیک در20-40% موارد +8,-7و ناهنجاریهای 12P می باشد، برخی از بیماران t(5,12) یا t(5,17) همراه با افزایش ائوزینوفیل وپاسخ درمانی مناسب به بازدارنده های تیروزین کینازی دارند. میزان متوسط بقا در بیماران ماه می باشد و درصد پیشرفت به سمت AML دارند.
 یا t(5,17) همراه با افزایش ائوزینوفیل وپاسخ درمانی مناسب به بازدارنده های تیروزین کینازی دارند. میزان متوسط بقا در بیماران ماه می باشد و درصد پیشرفت به سمت AML دارند.")
18
Atypical Chronic Myeloid Leukemia, BCR-ABL1 Negative
Criteria for diagnosis of aCML : Leukocytosis (WBC ≤13 × 109/L) due to increased neutrophils and precursors with prominent dysgranulopoiesis Prominent dysgranulopoiesis No Ph′ chromosome or BCR/ABL-1 fusion gene No rearrangement of PDGFRA or PDGFRB Neutrophil precursors (promyelocytes, myelocytes, metamyelocytes) ≥10% of WBCs Minimal absolute basophilia; basophils usually <2% of leukocytes No or minimal absolute monocytosis; monocytes <10% of WBCs Hypercellular marrow with granulocytic proliferation and granulocytic dysplasia, with or without dysplasia of erythroid and megakaryocytic lineages Less than 20% blasts in blood or marrow
 due to increased neutrophils and precursors with prominent dysgranulopoiesis. Prominent dysgranulopoiesis. No Ph′ chromosome or BCR/ABL-1 fusion gene. No rearrangement of PDGFRA or PDGFRB. Neutrophil precursors (promyelocytes, myelocytes, metamyelocytes) ≥10% of WBCs. Minimal absolute basophilia; basophils usually <2% of leukocytes. No or minimal absolute monocytosis; monocytes <10% of WBCs. Hypercellular marrow with granulocytic proliferation and granulocytic dysplasia, with or without dysplasia of erythroid and megakaryocytic lineages. Less than 20% blasts in blood or marrow.")
21
Atypical CML. Top panels: Peripheral blood smear from a patient with atypical CML showed a leukocytosis with anemia and marked thrombocytopenia. Granulocytes are left-shifted, with dysplastic hypolobulated nuclei and a dysplastic nRBC is present (arrow). Bottom panels: Biopsy at low- and high-magnification (left and right, respectively) showing atypical granulocytic hyperplasia without sheets of blasts.
 showing atypical granulocytic hyperplasia without sheets of blasts.")
22
Juvenile Myelomonocytic Leukemia
Juvenile myelomonocytic leukemia (JMML) is a clonal disorder of predominantly granulocytic and monocytic lineages, with dysplasia in these and frequently other lineages, occurring in children or young adolescents The occurrence is 1.3 cases per million children younger than 14 years, and most affected children are younger than 3 years of age, with a 2 : 1 male predominance. JMML is frequent in children with neurofibromatosis type 1.
 is a clonal disorder of predominantly granulocytic and monocytic lineages, with dysplasia in these and frequently other lineages, occurring in children or young adolescents. The occurrence is 1.3 cases per million children younger than 14 years, and most affected children are younger than 3 years of age, with a 2 : 1 male predominance. JMML is frequent in children with neurofibromatosis type 1.")
23
In all, 25% of patients show monosomy 7, 35% exhibit mutations of PTPN11 (encoding SHP2), and 20% each have mutations in NRAS, KRAS2, and NF1. Patients with normal karyotype often have markedly increased hemoglobin F. Blood shows leukocytosis, thrombocytopenia, and often anemia. The marrow is hyperplastic with granulocytic hyperplasia, variable erythroid cellularity, and often decreased megakaryocytes.

24
Criteria for diagnosis of JMML
Peripheral blood monocytosis >1 × 109/L Absence of Ph′ chromosome or BCR/ABL-1 Blasts and promonocytes less than 20% of blood and marrow Plus two of the following: Hemoglobin F increased for age Immature granulocytes in the blood WBCs >10 × 109/L Clonal chromosomal abnormality (may be monosomy 7) GM-CSF hypersensitivity of myeloid progenitors in vitro
 GM-CSF hypersensitivity of myeloid progenitors in vitro.")
25
This peripheral blood smear from a child with JMML shows dysplastic monocytosis, left-shifted leukocytosis with a small blast, and thrombocytopenia.

27
Myelodysplastic/Myeloproliferative Disease, Unclassifiable
This category of the WHO classification is used for those cases with features of myelodysplastic disease, but with the addition of prominent myeloproliferative features. Refractory Anemia with Ring Sideroblasts Associated With Marked Thrombocytosis

28
Refractory Anemia with Ring Sideroblasts Associated With Marked Thrombocytosis
This disorder is included in the WHO classification under the category of MDS/MPD, unclassified. It is characterized by features of myelodysplastic neoplasm and refractory anemia with ring sideroblasts. Peripheral thrombocytosis ≥450 × 106/L, and increased, large atypical BM megakaryocytes. A majority of cases (60%) carry a JAK2 V617F mutation identical to that in MPN; occasional cases show a MPL W515K/L mutation
 carry a JAK2 V617F mutation identical to that in MPN; occasional cases show a MPL W515K/L mutation.")
29
Myelodysplastic Syndromes
اختلالات کلونال خونسازی هستند. نارسایی BM علارغم سلولاریته بالای آن وجود دارد. از نظر شیوع در مطالعات اروپایی3-4 نفر در100000تا اما با افزایش سن افزایش می یابد بطوریکه 30 نفر در در سن بالای 80 سال میرسد نتایج در ایتالیا نشان داده که 60درصد مریضان سن بالای 70سال دارند. 20 درصد بیماران با CBC اتفاقی شناسایی می شوند.اما اغلب با تصویری از نارسایی BM شناسایی می گردند. شایعترین ناهنجاری کروموزومی در کروموزوم 5(FMS,FOS,RAS), 7(ERB-D), 8(MYC) که همگی Proto-oncogeneرا حمل می کنند، می باشد.موتاسیون RAS در 50% وموتاسیون FMS در 16% از MDSها گزارش شده است.
, 7(ERB-D), 8(MYC) که همگی Proto-oncogeneرا حمل می کنند، می باشد.موتاسیون RAS در 50% وموتاسیون FMS در 16% از MDSها گزارش شده است.")
31
The Who classification of MDS
Refractory cytopenia with unilineage dysplasia (anemia (RA),thrombocytopenia, or neutropenia) Refractory anemia with ringed sideroblasts Refractory cytopenia with multilineage dysplasia Refractory anemia with excess blasts (RAEB) 5q− syndrome Myelodysplastic Syndrome, Unclassified Childhood Myelodysplastic Syndrome; Refractory Cytopenia of Childhood
,thrombocytopenia, or neutropenia) Refractory anemia with ringed sideroblasts. Refractory cytopenia with multilineage dysplasia. Refractory anemia with excess blasts (RAEB) 5q− syndrome. Myelodysplastic Syndrome, Unclassified. Childhood Myelodysplastic Syndrome; Refractory Cytopenia of Childhood.")
32
Refractory Cytopenia with Unilineage Dysplasia
dysplasia affecting >10% of one myeloid cell lineage, and cytopenia of the affected cell line, with <10% dysplasia of other lineages. Del (20q), +8, abnormalities ofchromosome 5 or 7 Refractory Anemia Refractory Neutropenia Refractory Thrombocytopenia
, +8, abnormalities ofchromosome 5 or 7. Refractory Anemia. Refractory Neutropenia. Refractory Thrombocytopenia.")
33
RA: 5-10%از MDSها را تشکیل میدهد.
دیسپلازی مشخص وبارز مربوط به رده اریتروئیدی است. آنمی، رتیکلوسیتوپنی، اریتروسیتهای غیرطبیعی، دایمورفیک،آنیزوپویکیلوسیتوز خفیف تا مشخصی وجود دارد. درBM هایپرسلولاریتی معمول می باشد اما گاهی نرمو یا حتی هیپوسلولار می باشد.دیسپلازی اریتروئیدی از خفیف تا متوسط متغیر است. میزان بلاست در PB کمتر از 1% و در BM کمتر از 5% ومیزان سیدروبلاست حلقویBM کمتراز15% اریتروبلاستها می باشد. میزان بقا 66ماه و transformation به AML حدود 6 درصد می باشد.

35
Refractory Neutropenia
The absolute neutrophil count is <1.8 × 109/L. Toxic/secondary neutropenia is excluded. Dysplasia usually consists of nuclear hypolobation and hypogranulation. Refractory Thrombocytopenia Platelets are <100 × 109/L More than 10% of at least 30 megakaryocytes evaluated in smears and sections show dysplastic features of hypolobation, binucleation or multinucleation, and/or micromegakaryocytes.

36
Refractory Anemia with Ring Sideroblasts
Similar to RA with >15% ringed sideroblasts in BM Survival is similar to RA, with a lower progression to AL (≤2%).
.")
40
Refractory Cytopenia with Multilineage Dysplasia
PB with cytopenias of ≥2 cell lines, <1% blasts, <1 × 109/L monocytes. BM with dysplasia of ≥10% of precursors of ≥2 cell lines, <5% blasts Ring sideroblasts may be present; when numerous (>15%), the case is still classified as RCMD and is not categorized separately, as in prior classifications. Survival is ≈33 months, and AL conversion is 11%
, the case is still classified as RCMD and is not categorized separately, as in prior classifications. Survival is ≈33 months, and AL conversion is 11%")
41
RA and RARS, which infrquently show cytogenetic abnormalities (<10%)
RCMD and RCMD with ring sideroblasts (RCMD-RS) show cytogenetic abnormalities in up to 50%. These include trisomy 8, monosomy 7, del(7q), monosomy 5, del(5q), del(20q)
 show cytogenetic abnormalities in up to 50%. These include trisomy 8, monosomy 7, del(7q), monosomy 5, del(5q), del(20q)")
42
Refractory Anemia with Excess Blasts
RAEB-1: PB with <5% blasts, <1 × 109/L monocytes; BM with hypercellularity, dyspoiesis, 5%–9% blasts without Auer rods; Survival is usually less than 2 years(18month) in RAEB-1 with progressive marrow failure and cytopenias or progression to AL in 25% Clusters of 5–8 blasts and promyelocytes in the marrow interstitium are frequently seen on sections and have been referred to as abnormal localization of immature precursors(ALIP).
 in RAEB-1. with progressive marrow failure and cytopenias or progression to AL in 25% Clusters of 5–8 blasts and promyelocytes in the marrow interstitium are frequently seen on sections and have been referred to as abnormal localization of immature precursors(ALIP).")
43
Cases of RAEB with concurrent myelofibrosis are referred to as RAEB with fibrosis (RAEB-F). Megakaryocytes are increased and dysplastic, and reticulin fibrosis is significant, often resulting in a dry tap.

44
RAEB-2: PB with 5%–9% blasts, or 10%–19% BM blasts, or Auer rods Survival is usually less than 2 years(10 month) in RAEB-2 progressive marrow failure and cytopenias or progression to AL in 33% for RAEB-2. Cytogenetic abnormalities include +8, -5, del(5q), -7, del(7q), and del(20q)
, -7, del(7q), and del(20q)")
48
Myelodysplastic Syndrome with Isolated del(5q)
PB with thrombocytosis, <5% blasts; BM with increased, hypolobulated megakaryocytes, <5% blasts isolated deletion of chromosome region 5q. Some cases also exhibit a JAK2 V617F mutation.

49
5q-syndrome. blood smear showing macrocytic anemia and thrombocythemia
5q-syndrome. blood smear showing macrocytic anemia and thrombocythemia. Macrocytic RBCs with cellular diameters that exceed the nucleus of a small mature lymphocyte are seen.

50
5q-syndrome. Aspirate smear show increased numbers of the characteristic unilobular megakaryocytes.

52
Myelodysplastic Syndrome, Unclassified
The myelodysplastic syndrome, unclassified category is used when clinical and hematologic findings of myelodysplasia exist, but without specific features to allow placement in one of the other categories.

53
Childhood Myelodysplastic Syndrome; Refractory Cytopenia of Childhood
Persistent cytopenia with <5% blasts in the BM, dysplasia at least unilineage but usually multilineage, in the marrow and marrow is also frequently hypocellular. <2% in the blood, Pancytopenia is frequent, with macrocytic anemia showing anisopoikilocytosis. The cytogenetic abnormality of monosomy 7 is the most common genetic abnormality and is associated with progressive disease. Cases with trisomy 8 may show a long stable period.

54
This disorder may be morphologically indistinguishable from results of infection, vitamin deficiency (B12, folate, vitamin E), metabolic or autoimmune disease, paroxysmal nocturnal hemoglobinuria (PNH), or acquired aplastic anemia.
, metabolic or autoimmune disease, paroxysmal nocturnal hemoglobinuria (PNH), or acquired aplastic anemia.")
Similar presentations




























 Myelodysplastic / myeloproliferative diseases (MDS/MPD) >")

:>")
 leukemia Is characterized by an unregulated proliferation of myeloid elements in the bone marrow,>")


>")



