Download presentation
Presentation is loading. Please wait.

1
HEREDITARY ANEMIAS PROF. SAMIYA NAEEMULLAH Diplomate American Board of Pediatrics FAAP, FCPS Head of Pediatrics Department Islamic International Medical College

2
ANEMIA Defined as HB level below normal range Defined as HB level below normal range Varies with age and sex of the individual Varies with age and sex of the individual Neonate Hb < 14 g/dl Neonate Hb < 14 g/dl 1-12 months: Hb <10 g/dl 1-12 months: Hb <10 g/dl 1- 12 years Hb < 11 g/dl 1- 12 years Hb < 11 g/dl

3
IMPAIRED RED CELL PRODUCTION Congenital Red cell aplasia Congenital Red cell aplasia Diamond Blackfan Anemia Diamond Blackfan Anemia Fanconi Anemia Fanconi Anemia

4
INCREASED RED CELL DESTRUCTION Hemolytic Anemias Hemolytic Anemias RED CELL MEMBRANE DISORDERS Hereditary Spherocytosis RED CELL ENZYME DISORDER Glucose 6 – phosphate dehydrogenase deficiency (G6PD) Hemoglobinopathies Sickle cell disease Thalassemia
 Hemoglobinopathies Sickle cell disease Thalassemia")
5
LEARNING OBJECTIVES Discuss the etiology of different types of hereditary anemias? Discuss the etiology of different types of hereditary anemias? Differentiate between cell defects, enzymatic defects and hemoglobinopathies Differentiate between cell defects, enzymatic defects and hemoglobinopathies Be able to distinguish the morphology of different hemolytic anemias Be able to distinguish the morphology of different hemolytic anemias Discuss clinical features of sickle cell disease and Thalassemia Major Discuss clinical features of sickle cell disease and Thalassemia Major Correlate clinical features with pathophysiology of the disease Correlate clinical features with pathophysiology of the disease

6
DIAMOND BLACKFAN ANEMIA Etiology Etiology Autosomal dominant 15% Autosomal dominant 15% Autosomal recessive 15% Autosomal recessive 15% Spradic 80% Spradic 80% FH 20% FH 20%

8
ANEMIA

9
CONGENITAL ANOMALIES Short stature

10
DIAGNOSIS Low Hb% macrocyte Low Hb% macrocyte Low reticulocyte Count Low reticulocyte Count Normal bilirubin Normal bilirubin Absent red cell precussors on narrow Absent red cell precussors on narrow

11
INHERITED APLASTIC ANEMIAS Reduction or absence of all three main line in bone marrow leading to peripheral blood pancytopenia Reduction or absence of all three main line in bone marrow leading to peripheral blood pancytopenia

12
FANCONI’S ANEMIA Short stature Short stature Abnormal radii & thumbs Abnormal radii & thumbs Renal manifestation Renal manifestation Pigmented skin lesion Pigmented skin lesion Increased chromosomal breakage of peripheral blood lymphocytes Increased chromosomal breakage of peripheral blood lymphocytes

18
Schwachman Diamond Syndrome Schwachman Diamond Syndrome Autosomal Recessive disorder Autosomal Recessive disorder Bone marrow failure Bone marrow failure Pancreatic exocrine failure Pancreatic exocrine failure Skeletal abnormalities Skeletal abnormalities

19
RED CELL MEMBRANE DISORDERS Hereditary spherocytosis Hereditary spherocytosis Hereditary elliptocytosis Hereditary elliptocytosis

20
HEREDITARY SPHEROCYTOSIS Autosomal dominant Autosomal dominant No F/H – in 25% cases No F/H – in 25% cases Defect in genes for the skeletal proteins of the red cells membrane spectrin, ankyrin and band 3 red cell losses part of its membrane passing through spleen Defect in genes for the skeletal proteins of the red cells membrane spectrin, ankyrin and band 3 red cell losses part of its membrane passing through spleen

21
in surface to volume ratio – causes the cells to become spheroidal. in surface to volume ratio – causes the cells to become spheroidal. Less deformable than normal RBS Less deformable than normal RBS Destruction in micro vasculature of the spleen Destruction in micro vasculature of the spleen

23
Jaundice at birth Jaundice at birth Anemia Anemia Mild to moderate splenomegaly Mild to moderate splenomegaly Aplastic crises Aplastic crises Gall stones Gall stones

25
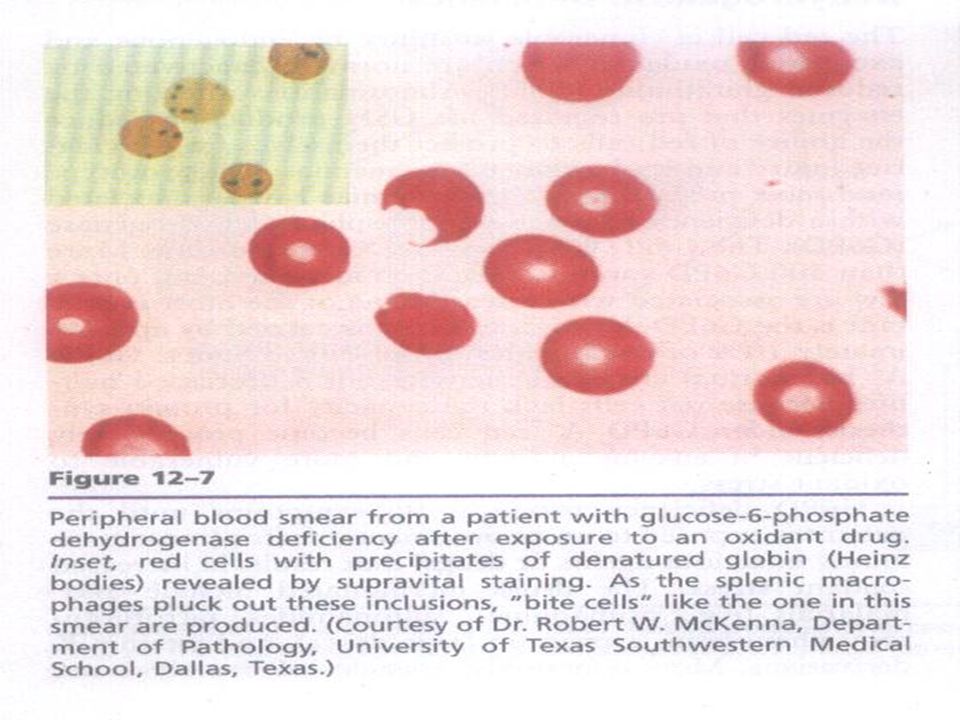
ENZYME DEFECTS Glucose 6 phosphate dehydrogenase (G 6 PD) deficiency Glucose 6 phosphate dehydrogenase (G 6 PD) deficiency Commonest red cell enzymopathy effecting over 100 million people world wide Commonest red cell enzymopathy effecting over 100 million people world wide G 6 PD in red cell essential for preventing G 6 PD in red cell essential for preventing Oxidative damage to red cells Oxidative damage to red cells So cell lacking the enzyme are suseptible to oxidant induced hemolysis So cell lacking the enzyme are suseptible to oxidant induced hemolysis
 deficiency Glucose 6 phosphate dehydrogenase (G 6 PD) deficiency Commonest red cell enzymopathy effecting over 100 million people world wide Commonest red cell enzymopathy effecting over 100 million people world wide G 6 PD in red cell essential for preventing G 6 PD in red cell essential for preventing Oxidative damage to red cells Oxidative damage to red cells So cell lacking the enzyme are suseptible to oxidant induced hemolysis So cell lacking the enzyme are suseptible to oxidant induced hemolysis")
27
Etiology with inherited as X linked effects males Etiology with inherited as X linked effects males G 6 PD activity in in Red cells G 6 PD activity in in Red cells Mostly in old cells Mostly in old cells

28
X LINKED INHERITANCE

29
CLINICALLY Neonatal Jaundice Neonatal Jaundice Acute hemolysis precipitected by Acute hemolysis precipitected by Drugs Drugs Infection Infection Naphthalene in moth balls Naphthalene in moth balls Intravascular hemolysis Intravascular hemolysis Fever, malaise, dark colored urine Fever, malaise, dark colored urine

30
HEMOGLOBINOPATHIES These are red blood cell disorders which cause hemolytic anemia because of These are red blood cell disorders which cause hemolytic anemia because of Reduced or absent production of Hb A( & thalassemia) Reduced or absent production of Hb A( & thalassemia) Production of abnormal Hb e.g. sickle cell disease Production of abnormal Hb e.g. sickle cell disease

31
BETA THALASSEMIA AUTOSOMAL RECESSIVE Inherited disorder characterized by absence or decreased synthesis of beta globin chain of hemoglobin Inherited disorder characterized by absence or decreased synthesis of beta globin chain of hemoglobin

33
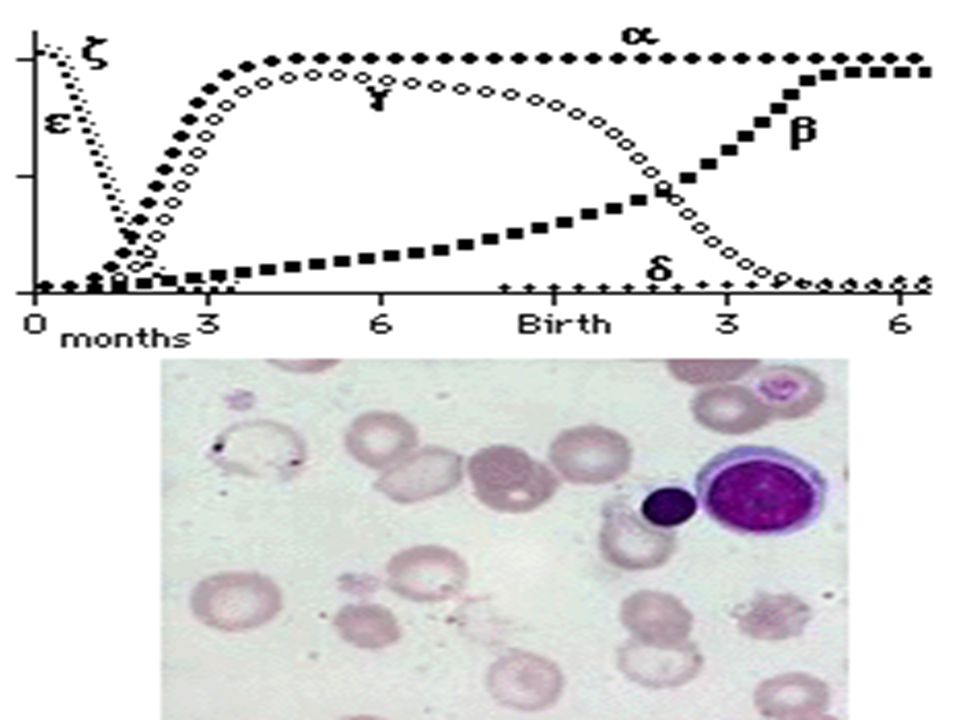
Normal Fetal Hb at different ages At birth – 70% At birth – 70% 5 weeks – 55% 5 weeks – 55% 4 months – 10% 4 months – 10% 5 month – 5% 5 month – 5% At one year – (< 2%) At one year – (< 2%)
 At one year – (< 2%)")
34
HETEROZYOUS SATE Thalassemia Minor One normal betaglobin chain gene and one beta-thalassemia gene One normal betaglobin chain gene and one beta-thalassemia geneHOMOZYGOUS Thalassemia Intermedia 2 Thalassemia genes 2 Thalassemia genes Thalassemia Major 2 Beta thalassemia genes 2 Beta thalassemia genes

35
PATHOPHYSIOLOGY Beta Thalassemia minor Most Common Most Common Failure of one gene coding for beta chain Failure of one gene coding for beta chain Alpha chain – production – normal Alpha chain – production – normal Alpha chain available combine with beta chain Alpha chain available combine with beta chain Hb A levels Hb A levels

36
Excess alpha chain – stimulates production of delta chain – Hb A 2 Excess alpha chain – stimulates production of delta chain – Hb A 2 Still excess alpha chains – switches off gamma chain production does not function correctly. And rate of gamma chain production is greater than in normal adult so amount of Hb F. Still excess alpha chains – switches off gamma chain production does not function correctly. And rate of gamma chain production is greater than in normal adult so amount of Hb F.

37
HbF Poor oxygen deliverer and high affinity for O 2 Poor oxygen deliverer and high affinity for O 2 Only functional Hb present is A 2 Only functional Hb present is A 2HypoxiaErythropoetin Stimulated marrow to maximum Extremedullary hematopaiesis Splenomegaly Splenomegaly

38
CLINICAL MANIFESTATIONS Thalassemia Minor The Growth & Development is normal The Growth & Development is normal Mild anemia Hb – 10 mg./dl Mild anemia Hb – 10 mg./dl

39
Thalassemia Intermedia Symptomatic by 2 – 4 yrs of age Symptomatic by 2 – 4 yrs of age Mod anemia Mod anemia Thalassemic facies Thalassemic facies Growth failure Growth failure Hepatosplenomegaly Hepatosplenomegaly Jaundice Jaundice

40
THALASSEMIA MAJOR DETROIT PEDIATRICIAN DETROIT PEDIATRICIAN 1925 THOMAS COOLEY (Cooley Anemia) 1925 THOMAS COOLEY (Cooley Anemia) Profound anemia Profound anemia Splenomegaly Splenomegaly Bony deformities Bony deformities Greek word Thalasa SEA Blood Greek word Thalasa SEA Blood
 1925 THOMAS COOLEY (Cooley Anemia) Profound anemia Profound anemia Splenomegaly Splenomegaly Bony deformities Bony deformities Greek word Thalasa SEA Blood Greek word Thalasa SEA Blood")
41
Universally fatal disease Is now converted into chronic illness

42
INCIDENCE 3% of world’s population carry Thalassemia gene 3% of world’s population carry Thalassemia gene PAKISTAN PAKISTAN Carrier rate 4-5% Carrier rate 4-5% Pathans 5.8% Pathans 5.8% Total No of patients 50,000-60,000 Total No of patients 50,000-60,000 5000 – 6000 children are born each year 5000 – 6000 children are born each year

45
THALASSEMIA MAJOR Born normal at birth Born normal at birth Starts getting pale,fussy,irritable Starts getting pale,fussy,irritable Starts refusing feeds Starts refusing feeds

46
INFANT

47
CHILD

48
CLINICAL FEATURE Bossing of skull Bossing of skull Maxillary overgrowth Maxillary overgrowth Long face Long face Hepatosplenomegaly Hepatosplenomegaly Bones become thin Bones become thin Fractures may occur Fractures may occur Heart failure Heart failure

49
ADOLESCENT

50
COMPLICATIONS Iron Overload Iron Overload Darkening of skin(Iron stimulated melanin) Darkening of skin(Iron stimulated melanin) Cardiomyopathy Cardiomyopathy Endocrinopathies Endocrinopathies Infections (Hepatitis A,B,C,Malaria,HIV) Infections (Hepatitis A,B,C,Malaria,HIV) Failure to thrive Failure to thrive Antibody formation(10%)Alloantibodies Antibody formation(10%)Alloantibodies
 Darkening of skin(Iron stimulated melanin) Cardiomyopathy Cardiomyopathy Endocrinopathies Endocrinopathies Infections (Hepatitis A,B,C,Malaria,HIV) Infections (Hepatitis A,B,C,Malaria,HIV) Failure to thrive Failure to thrive Antibody formation(10%)Alloantibodies Antibody formation(10%)Alloantibodies")
51
LAB DIAGNOSIS Complete Blood Count Complete Blood Count RBC Morphology RBC Morphology Hemoglobin Electrophoresis Hemoglobin Electrophoresis Serum Iron& Ferritin levels Serum Iron& Ferritin levels Imaging Study Imaging Study

55
IRON OVERLOAD LEAD TO Liver fibrosis Liver fibrosis Cardiomyopathy Cardiomyopathy Dysfunction of endocrine organs Dysfunction of endocrine organs Diabetes Diabetes Clinical Organ dysfunction Clinical Organ dysfunction Well transfused patient dies by 10-25 yrs if not chelated Well transfused patient dies by 10-25 yrs if not chelated

56
ADULT Fertility Ability to Reproduce and bare children Ability to Reproduce and bare children Reduce fertility due to iron overload Reduce fertility due to iron overload Control iron levels Pregnancy monitoring, reproductive assistance and perinotologist Pregnancy monitoring, reproductive assistance and perinotologist 40 cases reported

58
KEY MESSAGES Screen the Population Screen the Population Get The Blood Tested Get The Blood Tested Detect Thalessemia Traits Detect Thalessemia Traits Prevent them from marrying other thalassemia traits Prevent them from marrying other thalassemia traits Counselling Counselling Help the children & families with Thalassemia Help the children & families with Thalassemia

59
THALASSEMIA Healthy infants have four globin genes. Healthy infants have four globin genes. The manifestation of thalassemic syndromes depend on the number of functional globlin chains The manifestation of thalassemic syndromes depend on the number of functional globlin chains Deletion of 4 globin chains Deletion of 4 globin chains Hb Barts Hydrops fetalis Hb Barts Hydrops fetalis Fetal anemia – edema, ascities Fetal anemia – edema, ascities Fetus dies in utero or within hours of delivery Fetus dies in utero or within hours of delivery

61
Deletion of three globin genes Deletion of three globin genes (HbH disease) (HbH disease) Mild – moderate anemia Mild – moderate anemia Deletion of one or two globin chains Deletion of one or two globin chains thalassemia trait thalassemia trait Asymptomic Asymptomic Anemia is mild or absent Anemia is mild or absent
 (HbH disease) Mild – moderate anemia Mild – moderate anemia Deletion of one or two globin chains Deletion of one or two globin chains thalassemia trait thalassemia trait Asymptomic Asymptomic Anemia is mild or absent Anemia is mild or absent")
62
SICKLE CELL DISEASE HbSS Commonest genetic disorders in UK in 2000 births Commonest genetic disorders in UK in 2000 births Blacks from tropical Africa Blacks from tropical AfricaEtiology Point mutation in codon 6 of globin chain which causes change in aminoacid encoded from Point mutation in codon 6 of globin chain which causes change in aminoacid encoded from Glutamine to Valine Glutamine to Valine

63
FORMS Sickle cell anemia Sickle cell anemia SC disease SC disease Sickle thalassemia Sickle thalassemia

64
SICKLE CELL ANEMIA HbSS Patients are hemozygons for Hbs i.e. virtually all their Hb is Hbs Patients are hemozygons for Hbs i.e. virtually all their Hb is Hbs No HbA – no normal genes No HbA – no normal genes

65
SC DISEASE Affected children inherit Affected children inherit Hbs – one parent Hbs – one parent HbC- other parent HbC- other parent

66
SICKLE THALASSEMIA Affected children inherit Affected children inherit Hbs from – one parent Hbs from – one parent thalassemia – trait – 2 nd parent thalassemia – trait – 2 nd parent No normal -globin genes and most patients can make no Hb A so can transmit Hb S to their offspring No normal -globin genes and most patients can make no Hb A so can transmit Hb S to their offspring Symptoms are similar to those with sickle cell anemia. Symptoms are similar to those with sickle cell anemia.

67
SICKLE TRAIT Inheritance of HbS from one parent Inheritance of HbS from one parent Normal - globin gene – other parent Normal - globin gene – other parent 40% of the Hb is HbS 40% of the Hb is HbS Carriers of HbS Carriers of HbS Asymptomatic Asymptomatic

71
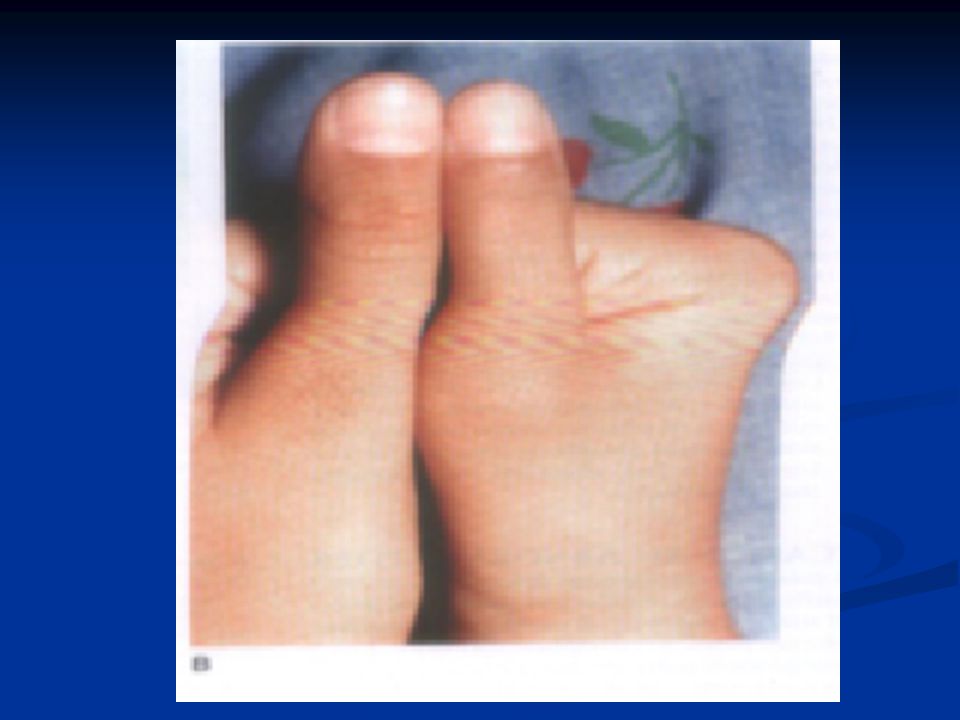
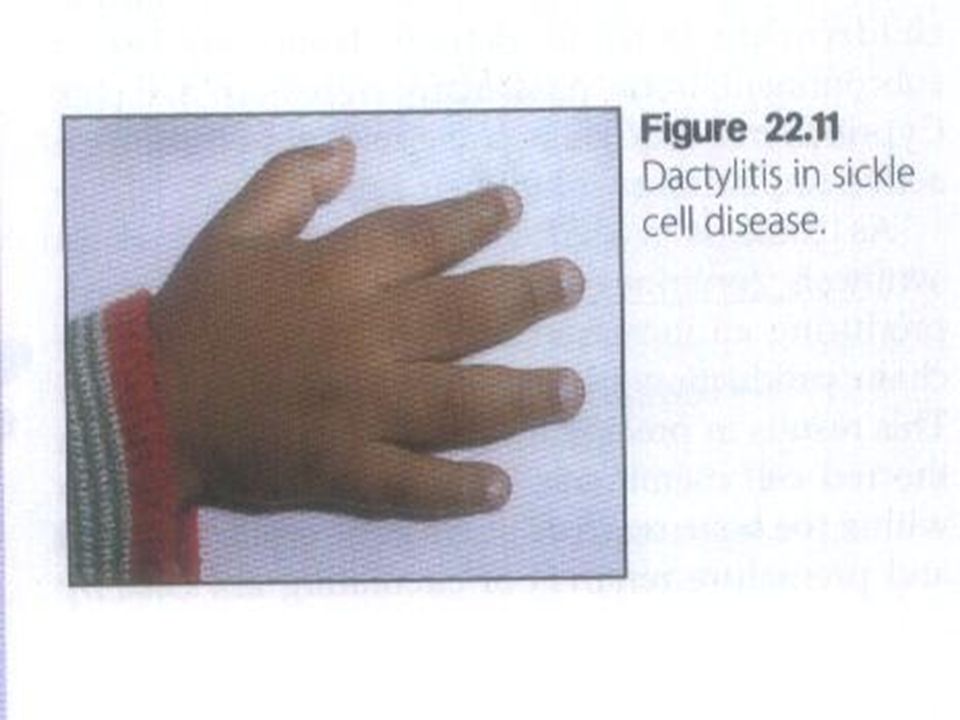
CLINICAL MANIFESTATIONS Anemia 6 – 8 gm/d Anemia 6 – 8 gm/d Jaundice Jaundice VASO - OCCLUSIVE CRISES Late infancy Hand – foot syndrome Hand – foot syndrome Bones of limbs & spine Bones of limbs & spine Cerebral & pulmonary infarction Cerebral & pulmonary infarction Acute stroke Acute stroke

73
PRECIPITATING FACTORS Exposure to cold Exposure to cold Dehydration Dehydration Excessive exercise Excessive exercise Stress Stress Hypoxia Hypoxia Infection Infection

74
ACUTE ANEMIA Hemolytic crisis Hemolytic crisis Associated with infection Associated with infection Aplastic crisis Aplastic crisis By parvo virus infection By parvo virus infection complete or temporary caestion of red blood cell production complete or temporary caestion of red blood cell production Sequestration crisis Sequestration crisis Sudden splenic enlargement Sudden splenic enlargement Abdominal pain Abdominal pain Circulatory collapse Circulatory collapse

75
INFECTIONS Susceptibily to infections as pneumococcus & hemophitus influenzae Susceptibily to infections as pneumococcus & hemophitus influenzae Increased incidence of Bone infection by salmonella Increased incidence of Bone infection by salmonella Chronic sickling microinfarction in spleen Chronic sickling microinfarction in spleen

76
Neonatal Diagnosis – at birth guthrie test Neonatal Diagnosis – at birth guthrie test Prenatal diagnosis by chorionic villus sampling at the end of 1 st trimester Prenatal diagnosis by chorionic villus sampling at the end of 1 st trimester

78
MANAGEMENT STEPS Regular red cell transfusions Regular red cell transfusions Chelation therapy Chelation therapy Growth monitoring and follow up Growth monitoring and follow up Management of complications Management of complications Psychosocial support Psychosocial support

79
Splenectomy Splenectomy Bone marrow transplantation Bone marrow transplantation Cord blood transplantation Cord blood transplantation Drugs stimulating gamma chains Drugs stimulating gamma chains Gene therapy Gene therapy Prevention and antenatal diagnosis Prevention and antenatal diagnosis MANAGEMENT STEPS

80
MULTI DISCIPLINARY APPROACH Pediatrician Pediatrician Hematologist Hematologist Gynecologist Gynecologist Physician Physician Surgeon Surgeon Ophthalmologist Ophthalmologist E NT Specialist E NT Specialist Lab technician Lab technician Psychologist Psychologist psychiatrist psychiatrist Social worker Parents Patient

Similar presentations






















 Hb is found in RBCs its main function is to transport O2 to tissues. Structure: 2 parts : heme + globin Globin: four globin chains (2 α.>")

 is composed of four protein chains, two α and two β globin chains arranged into.>")

. Red blood cells contain hemoglobin, an iron-rich.>")


 Hb is found in RBCs its main function is to transport O2 to tissues. Structure: 2 parts : heme + globin Globin: four chains. Heme: porphyrin.>")

i.>")



>")

