Download presentation
1
Histiocytoses

2
LANGERHANS CELL HISTIOCYTOSIS
. Letterer-Siwe disease . Hand-Schuller-Christian disease . Eosinophilic granuloma . Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker Disease)
")
3
Epidemiology most commonly develops in children ages
1-3 years, though disease can develop at any age. annual incidence of at least 5 per million children, with the adult incidence suspected to be less than one-third that of children. male:female ratio of nearly 2: I

4
Pathogenesis HHV-6 genetic basis elevated levels of cytokines

5
Clinical features Letterer-Siwe disease
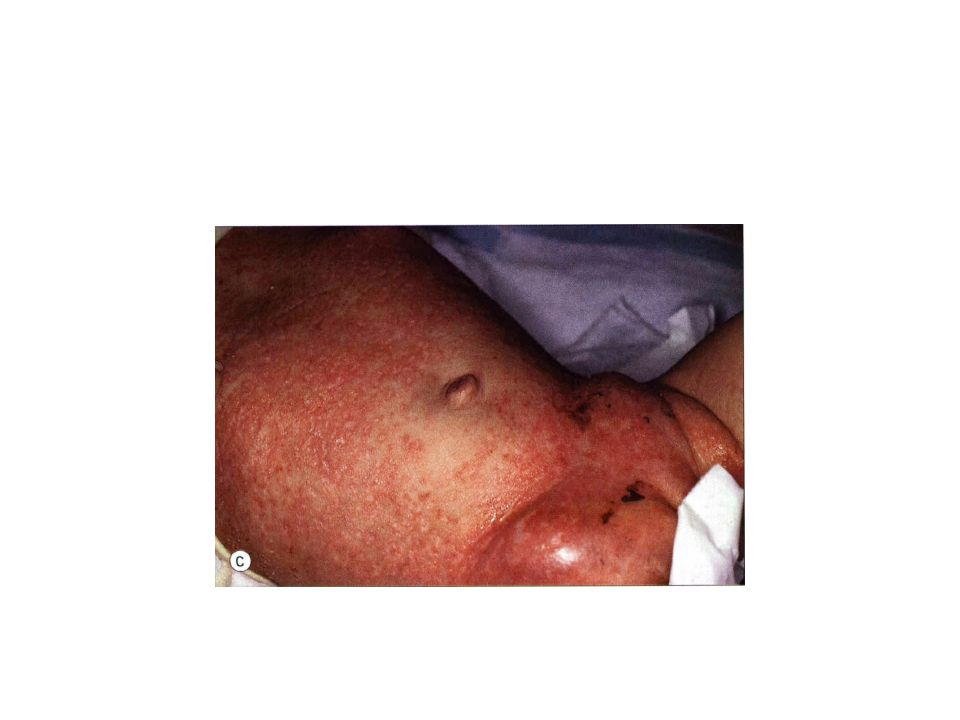
always develops prior to age 2 years, and commonly presents in children less than 1 year of age 1-2-mm pink to skin-colored papules, pustules and/or vesicles in the scalp, flexural areas of the neck, axilla and perineum, and on the trunk . The lesions tend to coalesce and become tender. Scale and crust with secondary impetiginization, and the development of petechiae and purpura are Comon Lung,liver, lymph node and bone involvement commonly occur at some point Occasionally, the hematopoietic system can be involved, with thrombocytopenia and anemia portending a poor prognosis.

9
Hand-Schuller-Christian
triad of diabetes insipidus, bone lesions and exophthalmous. begins between the ages of 2 and 6 years Approximately 30% of patients develop skin or mucous membrane lesions While early cutaneous lesions are similar to those seen in Letterer-Siwe disease, older lesions may become xanthomatousUlcerative nodules may develop in the oral and genitalareas, with premature loss of teeth possible secondary to gingival lesions. At least 80% of patients with Hand-Schuller-Christian disease develop bone lesions, the cranium being preferentially involved. Diabetes insipidus, secondary to infiltration of the posterior pituitary by LCH cells, develops in approximately 30% of patients

10
Eosinophilic granuloma
generally affects older children a single asymptomatic granulomatous lesion of the bone the most common manifestation . The cranium is most frequently affected.

12
Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease)
generally limited to the skin and rapidly self-healing. It presents at birth or in the first few days of life characteristic eruption of widespread red to brown papulonodules. After several weeks, the lesions crust and involute. Solitary papules, nodules and vesicles have also been observed Adults rarely develop LCH but, when they do, the most commonly involved sites are the skin, lung and bone.

14
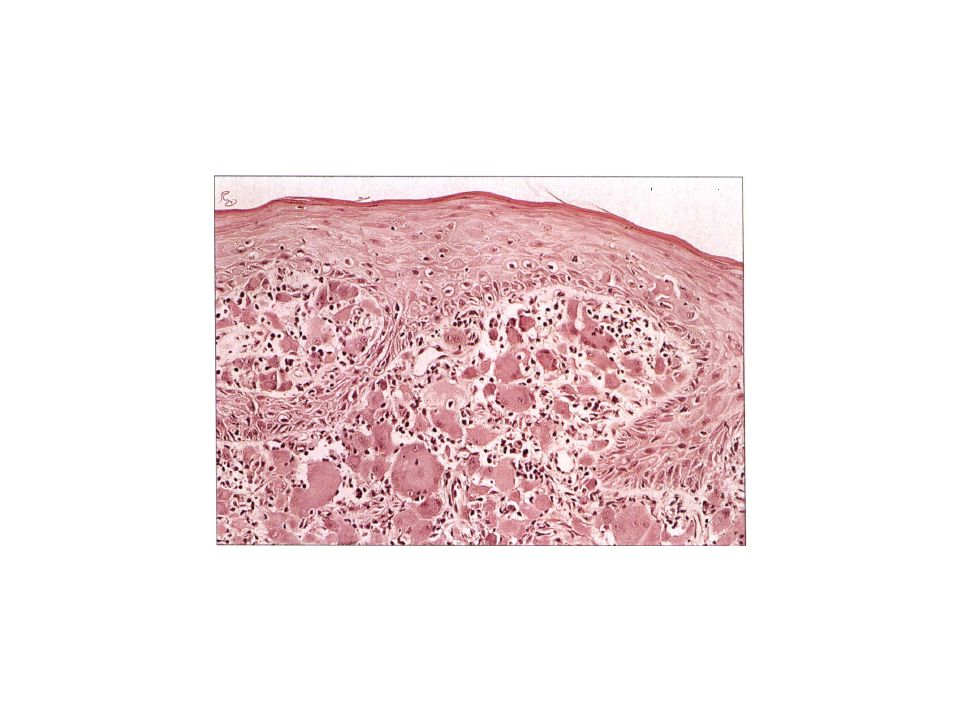
Pathology In a typical papule, a proliferation of LCH cells is present
in the papillary dermis . These cells are large, 10-15!lm in diameter, with a reniform (kidney-shaped) nucleus. LCH cells show positive immunostaining for CDla and SlOO ,Langerin (C0207) Electron microscopy demonstrates Birbeck granules, which are rod- or racquet-shaped cytoplasmic structures pathognomonic for Langerhans cells and LCH cells.
 nucleus. LCH cells show positive immunostaining for CDla and SlOO ,Langerin (C0207) Electron microscopy demonstrates Birbeck granules, which are rod- or racquet-shaped cytoplasmic structures pathognomonic for Langerhans cells and LCH cells.")
17
Treatment evaluation of the hematologic, pulmonary, hepatic, renal and skeletal systems For mild single-system skin disease (if treatment is required), topical corticosteroids, topical antibacterial agents, PUVA,and topical nitrogen mustard (mechlorethamine) have been reported to be effective in case Series For more extensive disease, thalidomide may be effective.
, topical corticosteroids, topical antibacterial agents, PUVA,and topical nitrogen mustard (mechlorethamine) have been reported to be effective in case Series. For more extensive disease, thalidomide may be effective.")
18
Benign Cephalic Histiocytosis
NON-LANGERHANS CELL HISTIOCYTOSES Benign Cephalic Histiocytosis

19
Epidemiology Pathogenesis
rare typically begins by the age of I year and always within the first 3 years of life. Pathogenesis given a similar histopathology, ultrastructural appearance and immunohistochemical profile to juvenile xanthogranuloma and generalized eruptive histiocytoma, several investigators have suggested that benign cephalic histiocytosis may represent a variant of juvenile xanthogranuloma or generalized eruptive histiocytoma.

20
Clinical features 2-5-mm, red to red-brown macules and papules, first on the face, with subsequent appearance on the ears and neck. The lesions spontaneously resolve after months or years. The papules flatten, become briefly hyperpigmented, and often disappear completely.

22
Pathology three histologic patterns in benign cephalic histiocytosis: a papillary dermal pattern, a diffuse pattern, and a lichenoid pattern. Treatment generally a self-limiting disorder and no treatment is needed.

23
Generalized Eruptive Histiocytoma
Epidemiology very rare Onset in adults is from the third to sixth decade. In children, onset is usually before age 4 years.

24
Clinical features recurrent crops of red to brown papules. At each occurrence, hundreds of papules, less than 1 cm, are distributed on the face, trunk and proximal extremities. In adults, there may be a symmetric arrangement of papules; mucosal surfaces are occasionally involved. Within several months, the lesions resolve completely or leave behind hyperpigmented macules or small Scars. Internal involvement has not been observed.

26
Pathology The superficial and mid dermis contain a nearly uniform infiltrate of histiocytes with a few lymphocytes. The histiocytes stain for lysozyme, ai-antitrypsin, CD II b,CD14b, Mac387, CD68 and factor XIIIa. Treatment No treatment is required as the disorder is self-limited.

27
Indeterminate Cell Histiocytosis Epidemiology
extremely rare The disorder occurs in adults, adolescents and infants, and a congenital form has also been reported. Pathogenesis The pathogenesis of indeterminate cell histiocytosis is unknown. However, it has most recently been speculated that indeterminate cells are dendritic cells en route from the skin to the regional lymph nodes.

28
Clinical features most cases of indeterminate cell histiocytosis have involved the trunk and extremities. Both a generalized form and a solitary form have been documented. In the generalized form, cutaneous lesions usually begin as firm red to brown papules, each less than 1 cm. In the solitary form, there is a single soft erythematous lesion, approximately 1cm in diameter. The course may wax and wane, though most patients experience a partial or complete regression of lesions. Mucous membrane involvement has not been observed. However, ocular involvement has been described, and visceral involvement and death have been reported in two instances.

29
Pathology monomorphous infiltrate of vacuolated! xanthomatized mononuclear histiocytes throughout the entire dermis. Lesional cells show expression of S100, CD la, HAM56, CD68, Mac387, lysozyme, ai-antitrypsin, HLA-DR, CDllc, CD14b and factor XIIIa. no Birbeck granules are found. Treatment Treatment of cutaneous lesions is not usually required as the condition is often self-limited and the lesions are asymptomatic. There are case reports of the beneficial effects of PUVA and 2-chlorodeoxyadenosine.

30
Juvenile Xanthogranuloma Epidemiology
fairly common disorder and the most common histiocytic disease of childhood. Almost 75% of cases appear during the first year of life, with over 15% being noted at birth. Juvenile xanthogranuloma is rare in adults, in whom its peak incidence is in the late twenties to early thirties. Pathogenesis The cause of juvenile xanthogranuloma is not known. however, it is often suggested that the condition is reactive with histiocytes possibly responding to a traumatic or infectious stimulus.

31
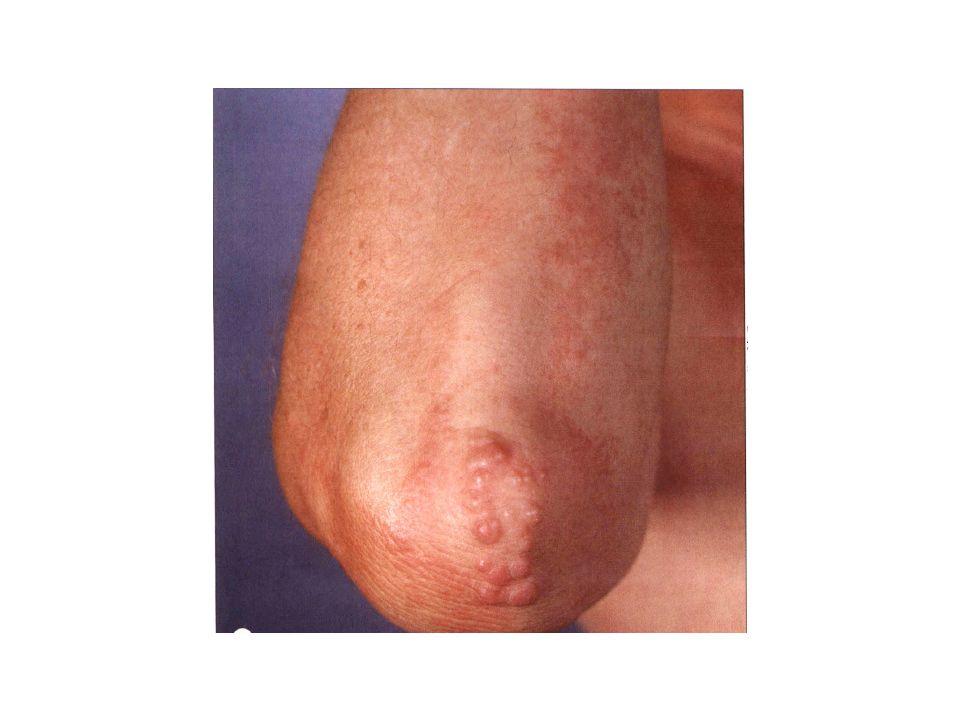
Clinical features two common clinical variants a small nodular form and large nodular form. Patients with the small nodular form, also known as the micronodular form, can present with many pink to red-brown, dome-shaped papules, 2-5 mm in diameter. The lesions are widely scattered on the upper part of the body and rapidly become yellow. the more common large nodular form is characterized by one or a few nodules 1-2 cm in diameter. Both forms frequently coexist. The most common location for juvenile xanthogranuloma is the head and neck ,followed by the upper torso, the upper extremities and the lower extremities.

32
Extracutaneous lesions have been reported in many organs,the eye being most commonly affected. Visceral, bone and CNS involvement is rare. The lung is the second most frequent extracutaneous site of disease. Hyphema (hemorrhage into the anterior chamber) and glaucoma are serious complications which can result in blindness. As well-recognizedassociation exists between juvenile xanthogranuloma and cafe-au-lait macules. 'triple association' consisting of juvenile xanthogranuloma, NF 1 and juvenile myelomonocytic leukemia. In the majority of patients with disease limited to the skin, the course is self-limited and benign. These patients are otherwise in good health and lesions usually regress within 3-6 years.
 and glaucoma are serious complications which can result in blindness. As well-recognizedassociation exists between juvenile xanthogranuloma and cafe-au-lait macules. triple association consisting of juvenile xanthogranuloma, NF 1 and juvenile myelomonocytic leukemia. In the majority of patients with disease limited to the skin, the course is self-limited and benign. These patients are otherwise in good health and lesions usually regress within 3-6 years.")
34
Pathology dense infiltrate of histiocytes in the superficial dermis in small lesions, extending into the subcutis in larger lesions. In the mature lesions, the histiocytes develop lipid in their cytoplasm, creating a foamy 'xanthomatous' appearance. Touton giant cells are a characteristic Finding stain positively for HAM56, CD68 and factor XIIIa. Some cases have shown expression of S100. CDla is usually negative. Treatment Due to the self-limiting nature of the eruption, no treatment is required.

36
Necrobiotic Xanthogranuloma
Epidemiology rare The average age of onset is the sixth decade Pathogenesis The strong association of necrobiotic xanthogranuloma with paraproteinemia has led to several hypotheses regarding the pathogenesis of necrobiotic xanthogranuloma. The paraprotein has been suspected either to be the primary inciting agent or to act as a cofactor in eliciting a giant cell granulomatous reaction.

37
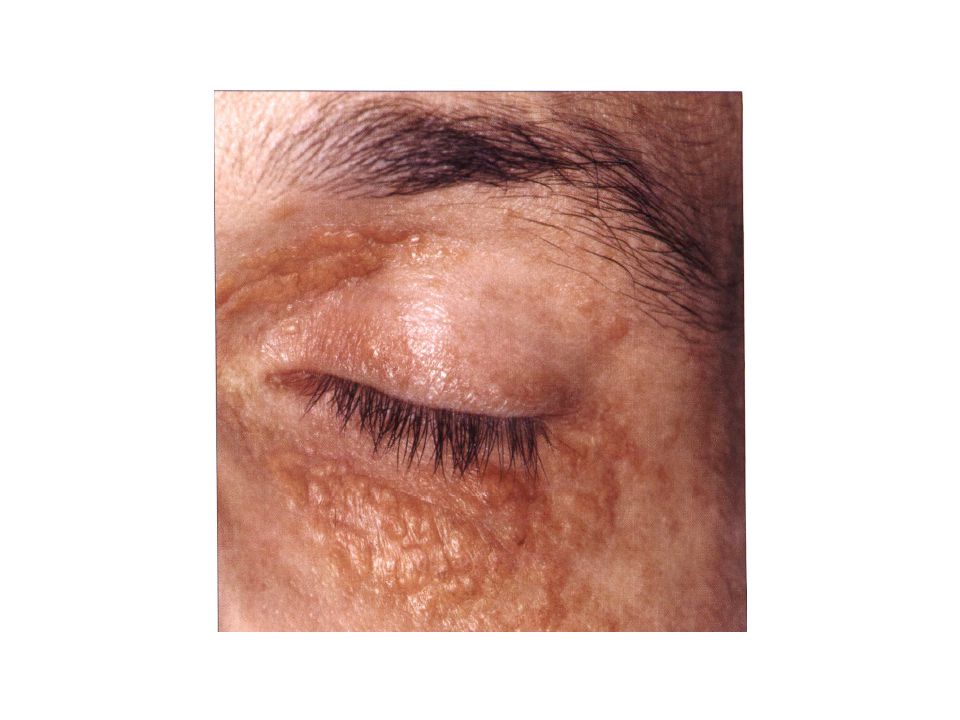
Clinical features The classic skin lesion is an asymptomatic indurated papule, nodule or plaque with a yellow 'xanthomatous' hue. Other features can include telangiectasias, atrophy, ulceration of lesions and scarring, with scars being common sites for development of new lesions. The periorbital region is the most common site of involvement. The trunk, remainder of the face, and proximal extremities are also frequently involved. Approximately 50% of patients have ophthalmic manifestations, which include orbital masses, ectropion, ptosis, conjunctival lesions, keratitis and scleritis, episcleritis, anterior uveitis, and proptosis.

38
A hallmark feature of necrobiotic xanthogranuloma is the associated paraproteinemia, an IgG monoclonal gammopathy, which is found in at least 80% of cases. Other common findings include hepatomegaly, splenomegaly, an increased ESR, leukopenia, hypocomplementemia, and underlying myeloma or plasma cell dyscrasia. Less commonly, cryoglobulinemia and/or an underlying Iymphoproliferative disorder are observed.

40
Pathology A palisading xanthogranuloma is found in the middle dermis
extending through the panniculus The granulomas consist of histiocytes, foam cells, lymphoid follicles, plasma cells and giant cells with zones of necrobiosis. Cholesterol clefts are found in areas of 'necrobiosis' (altered collagen, in which there appears to be necrosis,loss of collagen bundle integrity, and nuclear debris). A prominent feature is the presence of both Touton giant cells and large, bizarre foreign body giant cells. stain positively for lysozyme, CD68, Mac387 and CD lIb.
. A prominent feature is the presence of both Touton giant cells and large, bizarre foreign body giant cells. stain positively for lysozyme, CD68, Mac387 and CD lIb.")
42
Treatment No controlled clinical studies are available regarding the treatment of necrobiotic xanthogranuloma. Resolution or improvement of skin lesions has been seen in some patients treated with low-dose chlorambucil, melphalan or cyclophosphomide (systemic corticosteroids), radiation therapy, CO2 laser, and plasmapheresis.
, radiation therapy, CO2 laser, and plasmapheresis.")
43
Reticulo histiocytosis Epidemiology
All forms of reticulohistiocytoses occur predominantly in Caucasian adults. Pathogenesis The pathogenesis of the reticulohistiocytoses is unknown. However, it has been speculated that it represents an abnormal histiocytic response to various stimuli. Mycobacteria have been suggested as a possible trigger for multicentric reticulohistiocytosis. Others have suggested that the histiocytic response in multicentric reticulohistiocytosis is an immunologic process related to an underlying autoimmune or neoplastic disorder. The cause of giant cell retieulohistiocytoma is even less clear. Some lesions are believed to occur after trauma.

44
Clinical features Giant cell reticulohistiocytoma presents as a single, asymptomatic, yellow to red nodule. It can develop at any cutaneous site but is thought to favor the head. The patients are otherwise healthy and lesions tend to spontaneously resolve. Nodules are generally less than 1 cm. Multicentric reticulohistiocytosis is a disease characterized by cutaneous and mucous membrane reticulohistiocytomas and severe arthropathy. There is an association with hyperlipidemia, positive tuberculin skin test, systemic vasculitis, and autoimmune disease. Up to 28% of multicentric reticulohistiocytosis patients have reportedly had an associated malignancy, with bronchial, breast, stomach and cervical carcinomas being most common. An elevated ESR and anemia have been noted in approximately half of patients, and one-third demonstrate hypercholesterolcmia. Occasionally, IgG hypergammaglobulinemia and cryoglobulincmia have been observed. Fever and weight loss can occur.

45
Cutancous lesions range from a few millimetcrs to 2 cm and are
skin-colored to red, brown or yellow. Lesions tend to be acrally distributed. Favored sitcs include the head, hands, fingers, ears, and articular regions of the limbs . Small papules aligned alongthe periungual regions result in a characteristic 'coral bead' appearance. Approximately one-half of patients develop papules and nodules of the oral, pharyngeal and nasal mucosa. A 6-8-year course of symmetric, erosive arthritis of multiple joints is common and there is progression to arthritis mutilans in 45% of cases. The joints of the fingers and hands, as well as the knees and wrists, are most commonly involved. Rarely, there is histiocytic involvement of the heart, eye, lungs, thyroid, liver, kidney, muscle, salivary gland and or bone marrow. The disease spontaneously remits in 5-10 years, though patients are often left with significant disability.

48
Pathology dermal infiltrate of lymphocytes and histiocytes,with occasional plasma cclls and eosinophils. The histiocytes have a characteristic appearance. They are both mononuclear and multinuclear, with abundant eosinophilic, homogenous, and finely granular cytoplasm, creating a 'ground glass' effect. Giant cell reticulohistiocytoma and multicentric reticulohistiocytosis histiocytes stain positively for lysozyme and ai-antitrypsin and also express CD68, CDl1b, CD14 and HAM56. found that factor XIIIa was positive in cases of giant cell reticulohistiocytoma and negative in multicentric reticulohistiocytosis lesions.

50
Treatment Surgical excision of giant cell reticulohistiocytoma is curative. Most systemic therapy for multicentric reticulohistiocytosis has not been effective. Although not in controlled clinical trials, NSAIDs, oral corticosteroids, azathioprine, cyclophosphamide and chlorambucil have been tried, but with limited or no benefit. However, there are multiple case reports of substantial benefit with methotrexate, either alone or in combination with cyclophosphamide and corticosteroids.

51
Rosai-Dorfman Disease
(Sinus histiocytosis with massive lymphadenopathy) Epidemiology almost 600 cases had been reported by 1995. most commonly in children and young adults. Pathogenesis The etiology of Rosai-Dorfman disease has not been identified, though a viral pathogenesis has been postulated.EBV,HHV-6 More recently, it has been suggested that Rosai-Dorfman disease may be closely related to autoimmune lymphoproliferative syndrome, an inherited disorder associated with defects in Fas-mediated apoptosis
 Epidemiology. almost 600 cases had been reported by most commonly in children and young adults. Pathogenesis. The etiology of Rosai-Dorfman disease has not been identified, though a viral pathogenesis has been postulated.EBV,HHV-6. More recently, it has been suggested that Rosai-Dorfman disease may be closely related to autoimmune lymphoproliferative syndrome, an inherited disorder associated with defects in Fas-mediated apoptosis.")
52
Clinical features The characteristic clinical feature is massive, painless, bilateral cervical lymphadenopathy. elevated ESR and an IgG polyclonal hypergammaglobulinemia. A common finding is mild anemia. There are multiple reports of non-Hodgkin lymphoma occurring in association with Rosai-Dorfman disease. Immune disorders occur in approximately 15% of patients with Rosai-Dorfman disease. Anti-red blood cell autoantibodies and joint disease are the most common findings.

53
Over 40% of patients with Rosai-Dorfman disease have at least one extranodal site of involvement. The most common extranodal sites are the skin and soft tissue, the eyelid and orbit, the upper respiratory tract, major salivary glands, CNS and bone. Cutaneous involvement occurs in approximately 10% of cases. Cutaneous lesions are often multiple and appear as non-specific red to red-brown or xanthomatous macules, papules, nodules or plaque. The eyelids and malar regions are frequent sites of involvement.

55
Pathology dense dermal infiltrate of histiocytes with scattered lymphocytes, plasma cells and neutrophils.The histiocytes have large vesicular nuclei, small nucleoli, and abundant foamy, eosinophilic cytoplasm with feathery borders. Rosai-Dorfman disease histiocytes stain positively for S100, CD lIe,CD14, CD68, laminin 5 and lysozyme .Mac387 is occasionally expressed and examples positive for factor XIIIa have been reported.

57
Treatment Many lesions are asymptomatic and heal spontaneously, thus not requiring treatment. When treatment is indicated due to destructive lesions, disseminated disease or lesions causing physical compromise, radiotherapy, surgical excision, systemic corticosteroids, and alkylating agents have been used with some success.

58
Xanthoma Disseminatum Epidemiology
rare Age of onset ranges from 8 months to 85 years, though more than 60% of patients develop the disease before age 25 years. Pathogenesis The etiology of xanthoma disseminatum is unknown. As most patients have normal lipid levels, it has been suggested that xanthoma disseminatum represents a reactive proliferative disorder of histiocytes with secondary accumulation of lipid.

59
Clinical features Patients with xanthoma disseminatum may demonstrate the triad of cutaneous xanthomas, xanthomas of mucous membranes, and diabetes insipidus. The primary xanthomatous lesion is a yellow, red or brown papule. The onset of disease is marked by the eruption of hundreds of papules, symmetrically arranged, on the face and in the flexural and intertriginous areas of the trunk and proximal extremities The lesions tend to cluster into well-formed, potentially disfiguring Plaques. Mucous membrane lesions are found in 40-60% of patients with xanthoma disseminatum, the upper airway and oral mucosa being commonly involved.

60
Corneal and conjunctival lesions can threaten vision
Corneal and conjunctival lesions can threaten vision. CNS involvement of the hypothalamus and pituitary stalk results in diabetes insipidus in 40% of patients. Rare associations with xanthoma disseminatum have included plasma cell dyscrasia, monoclonal gammopathy and thyroid disorders. that patients follow one of three clinical courses: (1) a rare self-healing form with spontaneous resolution of lesions; (2) the common persistent form in which lesions may never resolve; and (3) the very rare progressive form with organ dysfunction and CNS involvement.
 a rare self-healing form with spontaneous resolution of lesions; (2) the common persistent form in which lesions may never. resolve; and (3) the very rare progressive form with organ dysfunction and CNS involvement.")
63
Pathology Fully developed lesions, however, demonstrate many foam cells and scattered histiocytes, lymphocytes, plasma cells, Touton cells and neutrophils. When intercellular accumulation of iron, or siderosis, is a prominent feature, the disorder has been called 'disseminated xanthosiderohistiocytosis'. siderosis is also found in juvenile xanthogranuloma . Xanthoma disseminatum histiocytes stain for lysozyme and aI-antitrypsin and also express CD68, CDllb, CD14, CDllc and factor XIIIa. Treatment For cutaneous and mucosal lesions, oral corticosteroids have not been of value, while the effectiveness of clofibrate has been mixed. Cyclophosphamide has been effective in the control of mucosal lesions. Cutaneous lesions have been treated hy CO2 laser, dermabrasion, radiotherapy, electrocoagulation, intralesional corticosteroids, cryotherapy and surgical excision.

























>")

>")









 to those.>")