Download presentation
Presentation is loading. Please wait.

1
Epi 260 Drug and Device Development What You Need to File an IND/IMPD April 11, 2012 Leslie Z. Benet, PhD Department of Bioengeering and Therapeutic Sciences Schools of Pharmacy and Medicine A good portion of the material presented in my presentation comes from the American Course in Drug Development and Regulatory Science (ACDRS) http://bts.ucsf.edu/ACDRS/
")
2
CONFLICT OF INTEREST STATEMENT Leslie Z. Benet, Ph.D. 7 / 11

3
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA FDA Home PageSearch FDA SiteFDA A-Z IndexContact FDA www.fda.gov FDA Organization FDA Overview

4
FDA Centers CBERCDERCDRHCFSANCVMNCTR Blood Products New Chemical & Biologic Drugs In vitro Diagnostics Domestic & Imported Food Safety Animal Drugs Biochemical & Molecular Markers VaccinesGeneric Drugs Medical Devices SeafoodMedicated Animal Feeds Quantitative Risk Assessment Cellular & Gene Therapy GMPs Radiological Products PesticidesGMPs Animal Bioassay Data Biological Establishments Labeling & Promotion GMPsCosmeticsIllegal Drug Use Caloric Restriction Tissues for Transplantation Pharmacokin- etic Research Device Standards Food & Color Additives Methods Development Neuro- toxicology Adverse Event Reporting Nutrition Labeling Developmental Toxicology Risk Assessment Methods Development

5
[www.fda.gov/Drugs/DevelopmentApprovalProcess/SmallBusinessAssistance/ucm053131.htm]www.fda.gov

7
Non-Clinical Components of an IND 1. Best available descriptive name of the drug, including the chemical name and structure of any new drug substance (NCE) 2.Complete list of components of the drug 3.Quantitative composition of the drug 4.Name and address of the supplier of any new drug substance and a description of the preparation (chemical synthesis or other method of manufacture) of any new drug substance
 2.Complete list of components of the drug 3.Quantitative composition of the drug 4.Name and address of the supplier of any new drug substance and a description of the preparation (chemical synthesis or other method of manufacture) of any new drug substance.")
8
Definition of a New Drug “…any drug that is not generally recognized as safe and effective under the conditions prescribed, recommended, or suggested in the labeling…” A drug may be considered “new” because of its composition, its use, its dosage, or its dosage form. It’s readily recognized that an NCE would be considered to be a new drug; however, a drug can be new resulting from composition of inactive ingredients, the proportion of ingredients, active or inactive, or the combination of ingredients, active or inactive

9
Non-Clinical Components of an IND 4.A description of the preparation (chemical synthesis or other method of manufacture) of any new drug substance Implications : Is the “drug” substance pure? Analytical methodology for drug substance and potential contaminants (intermediates) Validation of analytical methodology (Quality control vs. Quality assessment) Controls on chemical process development
 Validation of analytical methodology (Quality control vs. Quality assessment) Controls on chemical process development.")
10
Selecting the “right” physico-chemical form of the drug substance This process involves discovering, identifying and characterizing all available candidates (API [active pharmaceutical ingredient] polymorphic forms, solvates/hydrates and related salts, complexes/co-crystals etc), selecting the best crystal form(s) from the candidates, and determining process boundary conditions among the selected API form and other related forms, by thermodynamic principles.
![Selecting the right physico-chemical form of the drug substance This process involves discovering, identifying and characterizing all available candidates (API [active pharmaceutical ingredient] polymorphic forms, solvates/hydrates and related salts, complexes/co-crystals etc), selecting the best crystal form(s) from the candidates, and determining process boundary conditions among the selected API form and other related forms, by thermodynamic principles.](http://images.slideplayer.com/12/3581603/slides/slide_10.jpg "Selecting the right physico-chemical form of the drug substance This process involves discovering, identifying and characterizing all available candidates (API [active pharmaceutical ingredient] polymorphic forms, solvates/hydrates and related salts, complexes/co-crystals etc), selecting the best crystal form(s) from the candidates, and determining process boundary conditions among the selected API form and other related forms, by thermodynamic principles.")
11
Drug Synthesis Considerations Compound fully characterized Raw materials available Synthesis scaleable Impurity profile understood; identification of potential genotox impurities Sufficient quantities available for analytical development Final salt form resolution/crystallization defined pH and organic solvent solubility profile defined Reasonable COGs (cost of goods) possible
 possible")
12
Preliminary Testing of the Lead Compound Forced Degradation --Aids in the development of the analytical methods, especially specificity Preformulation and Excipient Compatability --Aids in the formulation approach and formulation possibilities “Open Dish” Stability --Aids in the selection of packaging and selection of expiry or retest dates

13
Non-Clinical Components of an IND 5. Methods, facilities and controls used for the manufacture, processing, and packaging of the new drug (here new drug is the product) Implications : Are the non-drug (excipients) pure? Analytical methodology for excipients and potential contaminants Validation of analytical methodology (Quality control vs. Quality assessment) Controls on chemical process development
 Implications : Are the non-drug (excipients) pure. Analytical methodology for excipients and potential contaminants Validation of analytical methodology (Quality control vs. Quality assessment) Controls on chemical process development.")
14
Non-Clinical Components of an IND 5. Methods, facilities and controls used for the manufacture, processing, and packaging of the new drug (here new drug is the product) Implications : Stability: effects of time (shelf life for drug, excipients and formulation), temperature, humidity SOPs (standard operating procedures) GMP (Good Manufacturing Practices)
 Implications : Stability: effects of time (shelf life for drug, excipients and formulation), temperature, humidity SOPs (standard operating procedures) GMP (Good Manufacturing Practices).")
15
Good Manufacturing Practices The requirement that drugs, and the methods used in, or the facilities or controls used in their manufacture, processing, packing, or holding conform with those practices that will assure that such drugs meet the requirements of the Act as to safety, and have the identity, strength, quality, and purity characteristics that they purport or are represented to possess. If they do not they are adulterated.

16
“Current good manufacturing practice” is not defined in the Act or the regulations. The regulations concern themselves with specific criteria for buildings, equipment, personnel, components, master formula and batch production records, production and control procedures, product containers, packaging and labeling, laboratory controls, distribution records, stability and complaint files.

18
FDA Statistics ~ 11,500 employees (October 1, 2009); ~ 60% in Centers; ~ 33% in Regulatory Affairs; ~ 7% in Commissioner’s Office Of all the money you spend 25¢ of every $1 goes to products regulated by the FDA Regulatory – 5 Districts responsible for > 135,000 establishments Budget FY 2007 $1.545 billion; $5/yr per person FY 2011 $2.447 billion; $7.85 yr person
; ~ 60% in Centers; ~ 33% in Regulatory Affairs; ~ 7% in Commissioner’s Office Of all the money you spend 25¢ of every $1 goes to products regulated by the FDA Regulatory – 5 Districts responsible for > 135,000 establishments Budget FY 2007 $1.545 billion; $5/yr per person FY 2011 $2.447 billion; $7.85 yr person")
19
Globalization of Drug Manufacturing In the United States: 80% of APIs imported 40% of finished drug products are imported India and China are primary sources of drugs imported into the US All drug makers must register with FDA, but FDA has limited resources for inspections The US market for imported drugs is growing

20
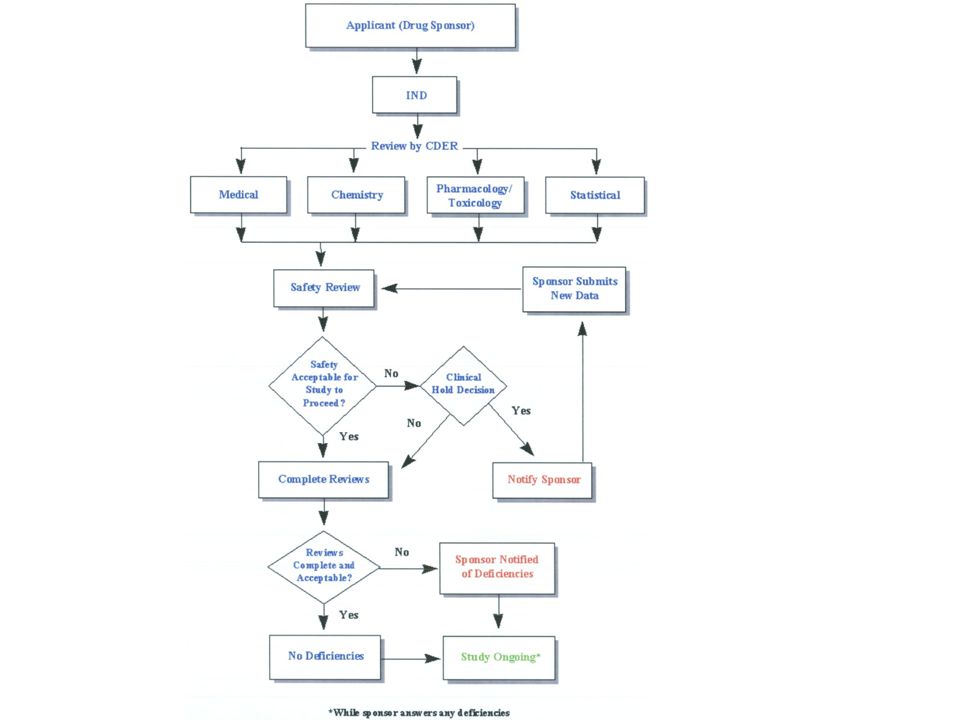
Components of an IND 6.A statement covering all information available to the sponsor derived from preclinical investigations and any clinical studies and experience with the drug. 7.Copies of the labels for the drugs and informational material that will be supplied to the investigators. This material must describe the preclinical studies with the drug and describe all relevant hazards, side effects, contraindications, and other information pertinent to use of the drug by the investigator.

21
Historical New Chemical Entity (NCE) Success Rate (Slide I first made about 20 years ago)
 Success Rate (Slide I first made about 20 years ago)")
22
Preclinical Animal Testing Tox & PK Toxicity Acute Tox, 2 weeks, rodent/non-rodent, male/female, single dose, dose rising Long Term Tox, 6 months, multiple dose, dose rising Mutagenicity Teratogenicity, rabbits Carcinogenicity, life time, rats PK/Metabolism Rodent/Non-rodent Male/Female Single dose, dose rising Multiple dose, dose rising Note—the preclinical exposure measurements set the upper limit for testing exposure in man

23
Metabolism Identify all the metabolites in each of the preclinical species (rat, mouse, dog, cyno) in liver microsomes, hepatocytes, whole animals and compare with human liver microsomes and hepatocytes (use in lead compound selection and comparison with competitors compounds) Concern with saturable metabolism; not so much with saturable absorption Inducing liver enzymes (usual kills a compound) Radiolabeled compound mass balance studies with lead compound.
 in liver microsomes, hepatocytes, whole animals and compare with human liver microsomes and hepatocytes (use in lead compound selection and comparison with competitors compounds) Concern with saturable metabolism; not so much with saturable absorption Inducing liver enzymes (usual kills a compound) Radiolabeled compound mass balance studies with lead compound.")
24
Pharmacokinetics Oral bioavailability Is clearance too high and half-life too short? Is clearance too low and half-life too long? Can animals be scaled to man? Time and dose nonlinearities (again increasing and decreasing dose normalized areas/exposure are worrisome) The extent of protein binding should have no influence on choosing a lead, except possibly for COG (cost of goods) considerations
 The extent of protein binding should have no influence on choosing a lead, except possibly for COG (cost of goods) considerations.")
25
Candidate 1 In Vivo DMPK Properties Clearance is high in rodents, moderate in dog and cyno Volume moderate (~1.5 L/kg) Half life short (~ 1 h) Mouse CL = 116 ml/min/kg Vss = 1.6 L/kg T1/2 = 0.75 h F = 27 % Fabs >1 Dog CL = 25 ml/min/kg Vss = 1.3 L/kg T1/2 = 0.75 h F = 82 % Fabs >1 Rat CL = 80 ml/min/kg Vss = 3.8 L/kg T1/2 = 0.5 h F = 57 % Fabs>1 Cyno CL = 15 ml/min/kg Vss = 1.3 L/kg T1/2 = 1.2 h F = 26 % Fabs 0.4 Bioavailability reasonable despite CL
 Half life short (~ 1 h) Mouse CL = 116 ml/min/kg Vss = 1.6 L/kg T1/2 = 0.75 h F = 27 % Fabs >1 Dog CL = 25 ml/min/kg Vss = 1.3 L/kg T1/2 = 0.75 h F = 82 % Fabs >1 Rat CL = 80 ml/min/kg Vss = 3.8 L/kg T1/2 = 0.5 h F = 57 % Fabs>1 Cyno CL = 15 ml/min/kg Vss = 1.3 L/kg T1/2 = 1.2 h F = 26 % Fabs 0.4 Bioavailability reasonable despite CL")
26
Candidate 2 In Vivo DMPK Properties Clearance is high in rat, moderate in dog and cyno Volume moderate (~1.5 L/kg) Half life short (~ 1 h) Bioavailability reasonable despite CL Good oral exposure in mouse Dog CL = 27 ml/min/kg Vss = 1.3 L/kg T1/2 = 0.7 h F = 64 % Fabs >1 Rat CL = 110 ml/min/kg Vss = 2.4 L/kg T1/2 = 0.5 h F = 40 % Fabs>1 Cyno CL = 21 ml/min/kg Vss = 1.9 L/kg T1/2 = 1.4 h F = 61 % Fabs >1
 Half life short (~ 1 h) Bioavailability reasonable despite CL Good oral exposure in mouse Dog CL = 27 ml/min/kg Vss = 1.3 L/kg T1/2 = 0.7 h F = 64 % Fabs >1 Rat CL = 110 ml/min/kg Vss = 2.4 L/kg T1/2 = 0.5 h F = 40 % Fabs>1 Cyno CL = 21 ml/min/kg Vss = 1.9 L/kg T1/2 = 1.4 h F = 61 % Fabs >1")
27
In Vivo Investigations – ABT Study CYP mediated metabolism is responsible for ~ 50% of the CL Evidence of non-CYP routes of elimination As CL is at or above rat liver blood flow this elimination may not be hepatic metabolic CL alone CL with CYP inhibitor Rat IV CL (ml/min/kg) 8841 G-589424
 8841 G")
28
Candidate 1 CL IVIVE Rodent clearance is under predicted from in vitro data Dog and Cyno clearance scale well from in vitro data Likely active renal elimination in rat Allometry predicts human CL of 4-11 ml/min/kg MouseRatDogCynoHuman Predicted CLhep Mics / Heps (ml/min/kg) 61 / 2829 / 1422 / 922 / 512 / 6 Predicted CLhep Mics / Heps using fub (ml/min/kg) 48 / 1722 / 817 / 712 / 48 / 5 Observed Total CL (ml/min/kg) (% Liver blood flow) 116 (>100%) 77 (>100%) 22 (71%) 15 (50 %) ~ 3 – 5 (15 - 25 %) Observed Renal CL (ml/min/kg) (% dose) 30 (20%) 1.2 (<5%) 1.3 (~8%) glomerular filtration rate (GFR) (ml/min/kg) 5-103-51-2 Max GFR*fu (ml/min/kg) (%PPB) 8 (20) 3.1 (38.2) 0.95 (52.5)
 61 / 2829 / 1422 / 922 / 512 / 6 Predicted CLhep Mics / Heps using fub (ml/min/kg) 48 / 1722 / 817 / 712 / 48 / 5 Observed Total CL (ml/min/kg) (% Liver blood flow) 116 (>100%) 77 (>100%) 22 (71%) 15 (50 %) ~ 3 – 5 ( %) Observed Renal CL (ml/min/kg) (% dose) 30 (20%) 1.2 (<5%) 1.3 (~8%) glomerular filtration rate (GFR) (ml/min/kg) Max GFR*fu (ml/min/kg) (%PPB) 8 (20) 3.1 (38.2) 0.95 (52.5)")
29
Candidate 1 Allometry CL 3.9 - 11 ml/min/kg Vss 1.25 – 1.5 L/kg Estimated from human PK: CL 3-5 ml/min/kg Vss ~ 1 L/kg

30
PhARMA established a Pharmaceutical Innovations Steering Committee (PISC) that attempted in a joint effort to assess the predictability of human PK from preclinical data and to provide a comparison of the available prediction methods in the literature. These results are presented in a series of five papers and a Lead Commentary in the October, 2011, volume 10, issue 10 of the Journal of Pharmaceutical Sciences Lead Commentary pp 4047-4049; Part 1 pp 4050- 4073; Part 2 pp 4074-4089; Part 3 4090-4110; Part 4 pp 4111-4126; Part 5 pp4127-4157.

31
Twelve PhARMA companies collectively provided nonclinical and FIH PK data on low molecular weight lead clinical candidate compounds (108 following oral administration in humans, of which 19 had also been given iv), exhibiting a wide array of physicochemical and structural properties. The data were anonymized by PhARMA prior to data analysis and the human data was withheld from the data analyst and only made available after all human predictions of human PK had been made. The analyst comprehensively all published methodologies on the same data set, a very onerous task. The 118 compounds were all under recent development, with all the necessary in vitro and in vivo data. (Class 1, 2, 3, 4: 15%, 57%, 12%, 23%)
.")
32
Selected Summary Although some prediction methods are better than others, there is currently no universal outstanding method. Prediction of human PK after iv dosing is much better than after oral administration Even the best methods could predict events after oral administration, within a factor of 2, only ~45% of the time, with a tendency to under predict AUC, probably primarily due to under prediction of F. Predictions for the Class 1 drugs were best. No one preclinical species was found to be superior to the others, nor were multiple species data shown to be superior to data from one species. PBPK on average was no better than allometry, and sometimes inferior

33
Formal (GLP) Preclinical Safety Testing General Toxicity and Tolerability (repeated dose) -- Two species (rodent and non-rodent) -- Broad and comprehensive assessment (screen) -- Duration at least as long as human exposure (min. 2 weeks) -- Define MTD (maximum tolerated dose), NOAEL (no-observed adverse effect level) and target organ/systems Safety Pharmacology -- Acute effects on vital integrated organ systems CNS (central nervous system) Respiratory Cardiovascular (HR, BP, rhythm) Genetic Toxicology -- Gene mutation (Ames, bacteria) -- Chromosome damage (mammalian cells) -- In vitro (target cells) and in vivo (target cells)
 -- Define MTD (maximum tolerated dose), NOAEL (no-observed adverse effect level) and target organ/systems Safety Pharmacology -- Acute effects on vital integrated organ systems CNS (central nervous system) Respiratory Cardiovascular (HR, BP, rhythm) Genetic Toxicology -- Gene mutation (Ames, bacteria) -- Chromosome damage (mammalian cells) -- In vitro (target cells) and in vivo (target cells).")
34
Continued Nonclinical Safety Testing (Usually not done until after Phase 1/2 Human Studies) Reproductive and Developmental Toxicity Assessment -- Two species (rodent, rabbit, non-human primate) -- Concentrate on periods of extraordinary susceptibility Organogenisis (“teratogenicity”); roughly equivalent to second trimester Peri- and post-natal development; roughly equivalent to neonates and infants -- Purpose is to label for use in pregnancy and lactation -- Disruption of early development patterns: physical and cognitive Carcinogenicity -- Two species (rodent); generally life-time exposure paradigms -- Multiple interpretational difficulties; threshold considerations -- Purpose is to label for extraordinary risks Specific Populations -- Juvenile animal studies Purpose driven (pharmacology, pharmacokinetics, adult effects) Cardiovascular (HR, BP, rhythm)
 Reproductive and Developmental Toxicity Assessment -- Two species (rodent, rabbit, non-human primate) -- Concentrate on periods of extraordinary susceptibility Organogenisis ( teratogenicity ); roughly equivalent to second trimester Peri- and post-natal development; roughly equivalent to neonates and infants -- Purpose is to label for use in pregnancy and lactation -- Disruption of early development patterns: physical and cognitive Carcinogenicity -- Two species (rodent); generally life-time exposure paradigms -- Multiple interpretational difficulties; threshold considerations -- Purpose is to label for extraordinary risks Specific Populations -- Juvenile animal studies Purpose driven (pharmacology, pharmacokinetics, adult effects) Cardiovascular (HR, BP, rhythm)")
35
Safety Risk: Prolonged QT Interval Toursades de Pointes -- Polymorphic ventricular contraction (arrhythmia) -- Target risk of <1:10,000 – 100,000 -- Predicted by prolongation of QT interval on EKG -- Cause believed to be disruption of cardiac channels Preclinical assessment in intact, non-rodent animal model -- Dose causing a 10% or greater prolongation in corrected QT -- C max comparison to upper levels of human exposure In vitro assessment -- Generally focused on the IKr channel (rapid delayed potassium rectifier channel) -- Voltage clamp studies of fresh cardiomyocytes -- hERG: mammalian cell lines transfected with human ether-related aGOGO gene (IKr channel) -- EC 50 concentration (50% reduction in current) 10 μM
 -- Target risk of <1:10,000 – 100, Predicted by prolongation of QT interval on EKG -- Cause believed to be disruption of cardiac channels Preclinical assessment in intact, non-rodent animal model -- Dose causing a 10% or greater prolongation in corrected QT -- C max comparison to upper levels of human exposure In vitro assessment -- Generally focused on the IKr channel (rapid delayed potassium rectifier channel) -- Voltage clamp studies of fresh cardiomyocytes -- hERG: mammalian cell lines transfected with human ether-related aGOGO gene (IKr channel) -- EC 50 concentration (50% reduction in current) 10 μM")
36
Biologics (proteins, mAbs) Consistency of therapeutic supply is problematic -- Source, process, stability, demonstrated comparability Importance of species selection -- Equivalent biological/pharmacological activity -- Extrapolatable pharmacokinetics -- Technical feasibility -- Biological surrogates (BSUR) Immunogenic potential (antibody formation) -- Reduced therapeutic effect -- Abrogate effect of endogenous ligands -- Prolong elimination Inflammatory reactions -- Complex, poorly-understood, species dependent immune system -- Potential for cytokine “storm”
 Consistency of therapeutic supply is problematic -- Source, process, stability, demonstrated comparability Importance of species selection -- Equivalent biological/pharmacological activity -- Extrapolatable pharmacokinetics -- Technical feasibility -- Biological surrogates (BSUR) Immunogenic potential (antibody formation) -- Reduced therapeutic effect -- Abrogate effect of endogenous ligands -- Prolong elimination Inflammatory reactions -- Complex, poorly-understood, species dependent immune system -- Potential for cytokine storm")
37
IND (FDA) vs IMPD (EUA) They are generally very similar. However, the differences can be seen on a web presentation prepared by Boehringer Ingelheim entitled “Harmonization of IND and IMPD” http://www.pda.org/Presentation/2008ClinicalTrial s/Harmonization-of-IND-and-IMPD-.aspx

Similar presentations



 Karol Godwin DVM.>")














 clinical trials in healthy volunteer subjects Oncology Drug Advisory Committee.>")

