Download presentation
Presentation is loading. Please wait.

1
Complete and Incomplete Metamorphosis

2
What is metamorphosis? Metamorphosis refers to the way that certain organisms develop, grow, and change form. Metamorphosis actually means "change".

3
Jewish in Catholic Prague Sickly Lonely Perceived human beings as
Felt he was an outsider Jewish in Catholic Prague Sickly Lonely Perceived human beings as being trapped by authority in a hopeless world Became frustrated at having to support his family Had to work in a meaningless bureaucratic job where he was just another pencil pusher Took time away from his writing Franz Kafka

4
Two Types of Metamorphosis
COMPLETE METAMORPHOSIS - has FOUR stages. INCOMPLETE METAMORPHOSIS - has THREE stages (p.s. incomplete doesn’t mean not finished. It just means that the adult in complete metamorphosis is completely different from the larva. A nymph and its adult form are not completely different. They’re only a little different.)
")
5
4 STAGES OF COMPLETE METAMORPHOSIS
Egg Larva Pupa Adult

6
EGG The female lays eggs.

7
LARVA Larva hatch from the eggs. They do not look like adult insects. They usually have a worm-like shape. Caterpillars(毛虫), maggots (蛆虫), and grubs(蛴螬)are all just the larval stages of insects. Larvae molt their skin several times and they grow slightly larger.
, maggots (蛆虫), and grubs(蛴螬)are all just the larval stages of insects. Larvae molt their skin several times and they grow slightly larger.")
8
ADULT Inside the cocoon, the larvae change into adults. After a period of time, the adult breaks out of the cocoon.

9
INCOMPLETE METAMORPHOSIS
Let’s take a closer look at each stage!

10
3 STAGES OF INCOMPLETE METAMORPHOSIS
Egg Nymph Adult

11
EGG A female insect lays eggs. These eggs are often covered by an egg case which protects the eggs and holds them together.

12
NYMPH The eggs hatch into nymphs.
Nymphs looks like small adults, but usually don't have wings. Insect nymphs eat the same food that the adult insect eats. Nymphs shed or molt their exoskeletons (outer casings made up of a hard substance called chitin) and replace them with larger ones several times as they grow. Most nymphs molt 4-8 times.
 and replace them with larger ones several times as they grow. Most nymphs molt 4-8 times.")
13
ADULT The insects stop molting when they reach their adult size. By this time, they have also grown wings.

14
ENDOCRINE- describing or relating to any gland or other group of cells that synthesizes hormones and secretes them directly into the blood, lymph, or other intercellular fluid Endocrine cells release protein and non-protein hormones Synthesis of hormones is orchestrated by the CNS Hormones effects are tissue dependent

15
Hormonal Control of Insect Metamorphosis
Temperature, Light, Stress, etc. Brain Prothoracicotropic促前胸腺hormone (PTTH) Corpus Allatum咽侧体 Prothoracic Gland前胸腺 Juvenile Hormone (JH)保幼激素 Ecdysteroid蜕皮激素 Pupa Adult Larva
 Corpus Allatum咽侧体. Prothoracic Gland前胸腺. Juvenile Hormone (JH)保幼激素. Ecdysteroid蜕皮激素. Pupa. Adult. Larva.")
16
Control of Metamorphosis by Internal and External Factors
Temperature, Light, Stress, etc. Brain Temperature (day degrees) Critical size matched (availability of food) Light (photoperiod) Chemicals Amount of moisture(湿度) Stress: mutagens, predators(肉食者), etc.
 Critical size matched (availability of food) Light (photoperiod) Chemicals. Amount of moisture(湿度) Stress: mutagens, predators(肉食者), etc.")
17
Ecdysone: “Molting Hormone”
Steroid hormone produced by prothoracic gland (lipid soluble, passes through cell membrane to the nucleus) Activates early response genes (TFs) and then late response genes (may cause differentiation,cell proliferation and migration, structural changes, apoptosis) Primes insect to respond to second hormone, EH EcR Ec Early Response: Transcription Factors Late Response: Transcription initiated by Transcription Factors USP Binding Site
 Activates early response genes (TFs) and then late response genes (may cause differentiation,cell proliferation and migration, structural changes, apoptosis) Primes insect to respond to second hormone, EH. EcR. Ec. Early Response: Transcription Factors. Late Response: Transcription initiated by Transcription Factors. USP. Binding Site.")
18
Chromosome Puffing in Flies
Observed in giant salivary gland chromosomes (no cell division after replication) Can be inhibited by actinomycin(放线菌素D) Puffing is where transcription is occurring. Ecdysone can be detected by fluorescent antibodies localized to the puffing Early puffs and late puffs seen in larva to pupa and pupa to adult molt
 Can be inhibited by actinomycin(放线菌素D) Puffing is where transcription is occurring. Ecdysone can be detected by fluorescent antibodies localized to the puffing. Early puffs and late puffs seen in larva to pupa and pupa to adult molt.")
19
Alternative Splicing of Ecdysone Receptor Pre-mRNA Creates Several Forms of the Ec-Receptor Allowing Cell Type Specific Ecdysone Response

20
Regulation of JH Levels
Juvenile Hormone Ecdysone Amount of Hormone Larva Pupa Adult Low = larva stage; Medium JH levels = pupa stage; No JH = adult stage Rate of release limited by synthesis Amounts of JH also regulated by protein degradation and methyltransferase levels (can be protected by JH binding proteins, degraded by JH esterase)
")
21
Metamorphosis in Action: Remember Imaginal Discs?
Adult Insect: - JH Immature Insect: + JH

22
Insect control by targeting metamorphosis
Juvenile hormone mimic: Keep insects in larval stage -- Effective control for insects such as mosquitoes Juvenile hormone antagonist: Cause death of larva or early metamorphosis -- Effective control for crop pests such as hornworm Genes for juvenile hormone binding hormone and JH esterase have been identified

23
Frogs: Tadpole to Adult

24
Hormonal Control of Frog Metamorphosis
Secretions of two hormones, thyroxine (T4) and triiodothyronine (T3) cause metamorphic changes Hormones have different effects depending on location in body Timing of changes regulated by tissue dependent hormone sensitivity Thyroid receptor is transcriptional repressor until thyroid hormone binds causing it to become a transcriptional activator Positive feedback loop is established between thyroid hormone and pituitary gland allowing incremental increases in hormone concentration
 and triiodothyronine (T3) cause metamorphic changes. Hormones have different effects depending on location in body. Timing of changes regulated by tissue dependent hormone sensitivity. Thyroid receptor is transcriptional repressor until thyroid hormone binds causing it to become a transcriptional activator. Positive feedback loop is established between thyroid hormone and pituitary gland allowing incremental increases in hormone concentration.")
25
Metamorphosis Transcriptional Activation Pituitary 垂体 Thyroid
Thyroid Hormones (T3 and T4)
")
26
TR RXR HIGH LOW Number of Receptors in Affected Tissue
Amount of Hormone LOW HIGH TR T3 Early Response: Transcription Factors Late Response: Transcription initiated by Transcription Factors RXR Binding Site

27
TH does not determine the developmental program, but initiates it
Changing the location of tissue or organ does not alter its response to TH Transplant eye to tail region Differentiates & grows into eye in response to TH while tail regresses Transplant tail to trunk Tail regresses while limb grows

29
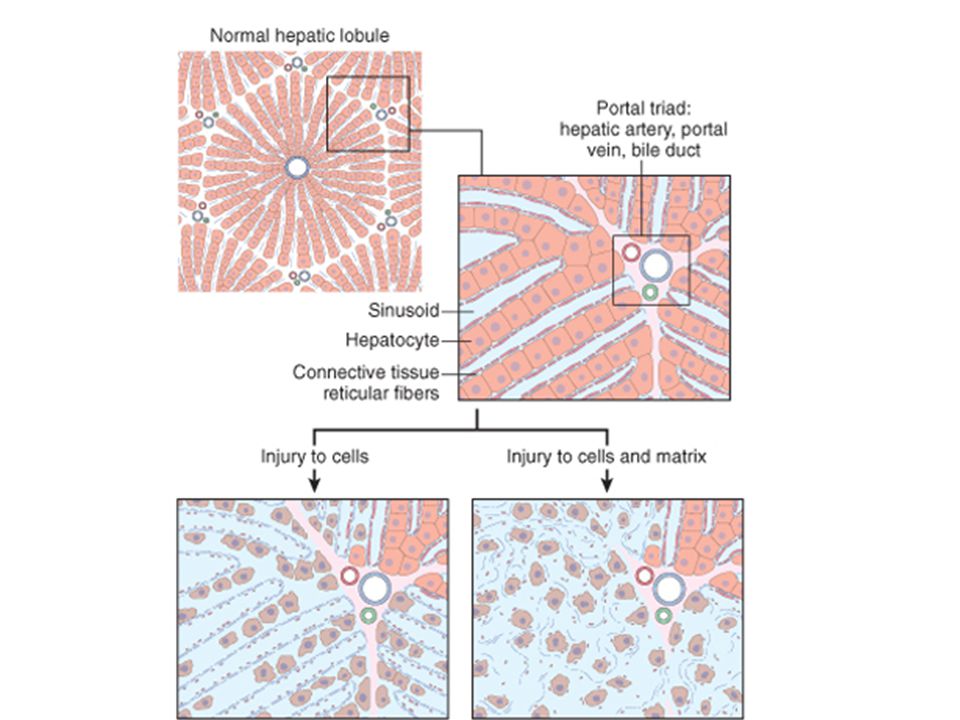
Regeneration, repair & healing of injured tissues
Tissue Repair Regeneration, repair & healing of injured tissues

30
Regeneration Growth of cells and tissues to replace lost structures
Requires an intact connective tissue scaffold

31
Regeneration of vertebrates
There are two types of regeneration: Epimorphosis or epimorphic regeneration : This type of regeneration involve the reconstruction of the missing parts by local proliferation from the blastema, or addition of parts to remaining piece . For example: regeneration of tail, limbs and lens in anurans and urodels and other vertebrates. 2. Morpholaxis or morphollactic regeneration: This type of regeneration involving reorganization of the remaining part of the body of an animal.For example: Hydra, planaria and other invertebrates e.g. regeneration of the new individual from body pieces.

32
Regeneration of Limb Regeneration begins in 3 phases :
Phase of wound healing or pre -blastema stage : Blood clotting and migration of epidermal cells from the basal layer of epidermis toward the centre of the wound. The wound is covered with epithelium which is thicker than the epidermis of the limb . 2. Phase of blastema formation : Cells accumulate beneath the epithelial covering and formed the blastema. Mesenchymal cells accumulate beneath the cap . Mesenchymal – blastemal cells differentiate into myoblasts and muscle cells, early cartilage cells and cartilage. During the dedifferentiate phase Hyaluronate (HA透明质酸) increases in the distal stump to form blastema . As the blastema forms, the HA will be decrease. The production of HA and break down of collagen represent the establishment of migration from stump tissues . 3. Phase of dedifferentiate and morphogenesis : The blastema begins to restore the part of which the limb was deprived. Specifically, if the fore arm is removed, the blastema differentiated directly into the muscle, bone, cartilage and skin of the fore arm.
 increases in the distal stump to form blastema . As the blastema forms, the HA will be decrease. The production of HA and break down of collagen represent the establishment of migration from stump tissues . 3. Phase of dedifferentiate and morphogenesis : The blastema begins to restore the part of which the limb was deprived. Specifically, if the fore arm is removed, the blastema differentiated directly into the muscle, bone, cartilage and skin of the fore arm.")
37
Tissue response to injury
Tissue response to injury. Repair after injury can occur by regeneration, which restores normal tissue, or by healing, which leads to scar formation and fibrosis. Tissue response to injury. Repair after injury can occur by regeneration, which restores normal tissue, or by healing, which leads to scar formation and fibrosis.

40
TYPES OF CELLS Labile cells Stable cells Permanent cells

41
Labile cells have a high rate of loss and replacement and therefore high capacity for regeneration. squamous and glandular epithelia haemopoeitic cells in bone marrow

42
Stable cells do not normally proliferate but can be stimulated to do so after damage. renal tubular cells, hepatocytes, osteoblasts, endothelial cells, fibroblasts.

43
Permanent cells Permanent cells : unable to divide after initial development and therefore cannot regenerate when some are lost. Neurons Skeletal & cardiac muscle

44
Repair Involves Regeneration of injured tissue by parechymal cells of the same type Replacement by connective tissue (fibrosis), resulting in a scar In most cases tissue repair involves both of these two processes.
, resulting in a scar. In most cases tissue repair involves both of these two processes.")
45
Repair by Connective Tissue (Fibrosis)
Fibrosis consists of four components formation of new blood vessels (angiogenesis) migration and proliferation of fibroblasts deposition of ECM maturation and reorganization of the fibrous tissue (remodeling)
 migration and proliferation of fibroblasts. deposition of ECM. maturation and reorganization of the fibrous tissue (remodeling)")
46
Repair responses after injury and inflammation
Repair responses after injury and inflammation. Repair after acute injury has several outcomes, including normal tissue restitution and healing with scar formation. Healing in chronic injury involves scar formation and fibrosis (see text).
.")
47
Biology of Aging

48
Some age gracefully... Goldie Jean Studlendgehawn
born on November 21, 1945

52
Animal Models in Aging Short lives, experimental results collected quickly, or over many generations; Maintained easily and inexpensively, present less complex genetic or physiological systems than humans. Genetically engineered animal models for exploring the basic mechanisms involved in the aging Saccharomyces cerevisiae Caenorhabditis elegans ANIMAL MODELS USED IN AGING RESEARCH: Animals are used in aging research for the same reasons as in other types of research. Besides the obvious difficulties that would be associated with doing experiments on humans, animal models are selected because they have relatively short lives, allowing for experimental results to be collected quickly, or over many generations; can be maintained easily and inexpensively, and present less complex genetic or physiological systems than humans. Genetically engineered animal models are particularly invaluable for exploring the basic mechanisms involved in the aging process as well as in extending our understanding of diseases found in the older human population. Some species that are widely used include yeast, Saccharomyces cerevisiae. the nematode worm, Caenorhabditis elegans, the fruit fly, Drosophila melanogaster, mice and rats. Saccharomyces cerevisiae – although technically not an animal, yeast cells provide many advantageous characteristics useful in the study of aging mechanisms: The cells are easy to grow, many biological processes that occur in yeast also occur in animal cells, and genetically different strains of yeast have different, but still short, life spans. Drosophila melanogaster Mus musculus

53
Theories of Aging: Oxidative Damage Telomeres
Genetic Alterations with aging Mitochondrial Aging Other processes involved: Inflammatory processes Hormonal changes Life style choices

54
Oxidative stress: imbalance production/breakdown
Free radicals are normal products of metabolism Predominant cellular free radicals are: - superoxide (O2 -) - hydroxyl (OH-) - nitrogen dioxide (NO2) - hydrogen peroxide (H2O2) superoxide
 - hydroxyl (OH-) - nitrogen dioxide (NO2) - hydrogen peroxide (H2O2) superoxide.")
55
Oxidative stress can lead to:
Damage to mitochondria, DNA, protein processing and cellular metabolism - lipid peroxidation - protein oxidation - DNA oxidation This ultimately leads to: - Loss of cellular phenotype - Necrosis - or Apoptosis

56
Endogenous defense mechanisms:
H2O Mitochondria P450 oxidases Catalase H2O2 O2- SOD OH+ H2O Superoxide dismutase and glutathione function to reduce oxide and peroxide, respectively, to harmless water. These two enzymes are reduced with aging, and in specific diseases such as Parkinson’s disease and Alzheimer’s disease. Too much SOD can also be damaging, as it leads to overproduction of peroxide, without the possibility to continue breakdown by glutathione. This is one hypothesis for what may go wrong in individuals with Down’s syndrome, who have an overproduction of SOD but maybe lower levels of glutathione in certain regions of the brain. Superoxide: The six outer shell electrons of each oxygen atom are shown in black; one electron pair is shared (middle); the unpaired electron is shown in the upper left and the additional electron conferring a negative charge is shown in red. Iron (Fe++) is converted to Fe+++ in the step between H2O2 and OH Glutathione is the most abundant intracellular nonprotein thiol and as such plays an important role in scavening cellular free radicals along with the enzyme glutathione peroxidase (GluPx). GSH is converted to GSSG or GSSCy during this process and is reduced back to GSH by the enzymatic activity of glutathione reductase. Studies have shown that GSH content decreases with age in many tissues including the brain. Glutathione plays an important role in brain by removing ROS formed during normal cellular metabolism such as during oxygen utilization by the mitochondria. GSH is synthesized by a twostep reaction involving the enzymes gammaglutamyl cysteine synthetase GCL, and glutathione synthetase, GS. GSH is synthetized in the cytosol and transported into the mitochondria via an energy dependent transporter. Decreases in GSH availability in the brain is believed to promote mitochondrial damage most likely via incresaes in levels of oxidative stress in this organelle. The magnitude of GSH loss seems to parallel the loss of DA neurons in PD and is the earliest known change, earlier than reported mitochondrial Complex 1 activity loss and striatal DA loss. GSH administration to a small group of PD patients via intravenous treatment over a month was reported to result in significant improvement of disability suggesting that GSH depletion may be a possible target for the disease. USE THIS FOR SPECIFIC ACTIVITY later Antioxidant scavengers: Glutathione Ascorbate (Vitamin C; inhibits oxidation) Vitamin E (a-tocopherol; inhibits lipid peroxidation) Carotenoids, flavonoids (present in brain??) Antioxidant enzymes: - Cu/Zn superoxide dismutase (SOD-1) - Mn superoxide dismutase (SOD-2) These enzymes catalyze the conversion of O2- to H2O2. H2O2 is then converted to H2O by glutathione peroxidase. 2 GSH Aging↓ Glutathione Reductase Glutathione Peroxidase GSSG H2O
; the unpaired electron is shown in the upper left and the additional electron conferring a negative charge is shown in red. Iron (Fe++) is converted to Fe+++ in the step between H2O2 and OH. Glutathione is the most abundant intracellular nonprotein thiol and as such plays an important role in scavening cellular free radicals along with the enzyme glutathione peroxidase (GluPx). GSH is converted to GSSG or GSSCy during this process and is reduced back to GSH by the enzymatic activity of glutathione reductase. Studies have shown that GSH content decreases with age in many tissues including the brain. Glutathione plays an important role in brain by removing ROS formed during normal cellular metabolism such as during oxygen utilization by the mitochondria. GSH is synthesized by a twostep reaction involving the enzymes gammaglutamyl cysteine synthetase GCL, and glutathione synthetase, GS. GSH is synthetized in the cytosol and transported into the mitochondria via an energy dependent transporter. Decreases in GSH availability in the brain is believed to promote mitochondrial damage most likely via incresaes in levels of oxidative stress in this organelle. The magnitude of GSH loss seems to parallel the loss of DA neurons in PD and is the earliest known change, earlier than reported mitochondrial Complex 1 activity loss and striatal DA loss. GSH administration to a small group of PD patients via intravenous treatment over a month was reported to result in significant improvement of disability suggesting that GSH depletion may be a possible target for the disease. USE THIS FOR SPECIFIC ACTIVITY later. Antioxidant scavengers: Glutathione. Ascorbate (Vitamin C; inhibits oxidation) Vitamin E (a-tocopherol; inhibits lipid peroxidation) Carotenoids, flavonoids (present in brain ) Antioxidant enzymes: - Cu/Zn superoxide dismutase (SOD-1) - Mn superoxide dismutase (SOD-2) These enzymes catalyze the conversion of O2- to H2O2. H2O2 is then converted to H2O by glutathione peroxidase. 2 GSH. Aging↓ Glutathione. Reductase. Glutathione. Peroxidase. GSSG. H2O.")
57
Telomeres and Aging Repetitive DNA sequences At the ends of all human
Chromosomes In humans there are 46 chromosomes; thus 92 telomeres (one per end) Telomere is about 10 to 15 kb in length, composed of the tandem repeat sequence: TTAGGG From: Aditya Rana in Biotechnology Shortened telomeres in mitotic cells may be responsible for some age-related changes (lack of plasticity) Telomerase can “fix” telomeres and “bypass” aging process Telomerase is a cellular reverse transcriptase, cellular immortalizing enzyme (germ cells and cancer cells) Adds TTAGGG repeats onto telomeric ends Cells in tissue culture can divide for 250 generations or more past the time they normally stop dividing when introducing telomerase, and are continuing to produce normal cells with normal amounts of chromosomes Most cells don’t have telomerase (cancer cells and reproduction cells and maybe also some stem cells) The telomerase control gene has been mapped to chromosome 3. Even though telomerase itself is present in most cells, the control gene is only present in cancer and reproductive cells (immortal cells)
 Telomere is about 10 to 15 kb in length, composed of the tandem repeat sequence: TTAGGG. From: Aditya Rana in Biotechnology. Shortened telomeres in mitotic cells may be responsible for some age-related changes (lack of plasticity) Telomerase can fix telomeres and bypass aging process. Telomerase is a cellular reverse transcriptase, cellular immortalizing enzyme (germ cells and cancer cells) Adds TTAGGG repeats onto telomeric ends. Cells in tissue culture can divide for 250 generations or more past the time they normally stop dividing when introducing telomerase, and are continuing to produce normal cells with normal amounts of chromosomes. Most cells don’t have telomerase (cancer cells and reproduction cells and maybe also some stem cells) The telomerase control gene has been mapped to chromosome 3. Even though telomerase itself is present in most cells, the control gene is only present in cancer and reproductive cells (immortal cells)")
58
Without telomeres, ends of chromosomes would be “repaired”, leading to chromosome fusion and abnormal function Telomeres regulate how many times a cell can divide. Telomere sequences shorten each time DNA replicates Once telomeres shrink to a certain level, cells can no longer divide; hence aging From: National Institutes on Aging (not protected by copyright); official domain
; official domain.")
59
Summary of Telomere theory:
Telomere length declines in dividing cells as we age 8,000 1,500 3,000 Age in years Telomere length in bp (human blood cells) With aging, the telomeres get shorter and shorter and when they reach a certain minimum size, the cell can no longer divide. Telomerase can “bypass” this process by repairing telomeres and adding the repeats. This phenomenon has not been shown in vivo yet; uncertain if it is also the cause for aging of the individual, not just the cells.
 With aging, the telomeres get shorter and shorter and when they reach a certain minimum size, the cell can no longer divide. Telomerase can bypass this process by repairing telomeres and adding the repeats. This phenomenon has not been shown in vivo yet; uncertain if it is also the cause for aging of the individual, not just the cells.")
60
Genes that affect Aging:
Stress resistance genes Genes targeting inflammation Genes that slow basic metabolism, like IGF Overall genetic stability The problem: Altered gene expression resulting from quality control defects allows errors to accumulate as cells divide leads to cells with diminished function Many scientists have hypothesized that some genes may control aspects of aging separate from the development of disease. These hypotheses are based on experimental studies of non-human organisms and the observation that longevity in humans appears to run in families. Studies of yeast and roundworm have identified over ten genes in each that are associated with longevity and aging, and more recent studies have suggested similar genes exist in the fruit fly. The exact function of these genes is unknown, but one or more may help slow down the metabolic rate. Studies in mice have shown that reducing metabolism by reducing food intake can increase life span. Finally, shortening of the telomeres decreases longevity in some model organisms. Finding similar genes in humans is more complicated, since scientists cannot experimentally control genes to test their effects on longevity in humans. Therefore, genetic studies of human longevity require a more observational approach. One study design is to examine large numbers of long-lived individuals such as centenarians and see what factors they have in common, such as lifestyle, medical history, and genetics. variants in multiple genes, including the human leukocyte antigen (HLA) genes of the immune system, apolipoprotein E (APOE), angiotensin-converting enzyme (ACE), plasminogen activating inhibitor 1 (PAI-1), and p53, are associated with living past age ninety. Forms of several of these genes, such as APOE, ACE, and p53, are associated with increased risk of developing Alzheimer's disease, cardiovascular disease, and cancer, respectively. The association of these genes with longevity may be due to these disease associations, or it may be due to their direct influence on extending the human life span. Regardless, genes clearly influence aging and longevity, whether it is by influencing the development of life span-shortening diseases, or by positively influencing longevity independently of causing disease. The receptors include those that are targeted by drugs used to treat high cholesterol and triglyceride levels. Mark Mattson et al One theory of aging assumes that the life span of a cell or organism is genetically determined—that the genes of an animal contain a “program” that determines its life span just as eye color is determined genetically, but what are the genes responsible? Nobody knows,… Error theory: Kiecolt-Glaser, 2003; Ly et al., 2000) Stress resistance genes: In model systems such as C Elegans, extended longevity is often associated with increased stress resistance (see eg. Longo, 1999). Manipulations in C elegans that extend longevity have a strong correlation to stress resistance, in the form of oxidative stress or inflammation probes. Cell lines from long lived species are stress resistant. Stress resistans in vitro also correlates with mamalian longevity (Kapahi et al., 1999). Cells taken from individuals with progeroid syndromes are more susceptible to stress (Gebhart et al., 1988). Genetic Expression - In certain instances, individual genes have been found that appear to directly affect longevity or rate of aging, but they do so through different mechanisms. Over expression of the urokinase plasminogen activator (uPA) in mice brains results in a 20% increase in longevity, probably by producing effects similar to Caloric Restriction. Urokinase plasminogen-activator is a serine protease that is important in embryonic development, aging and the onset of pathogenic conditions. Interestingly enough, deletion of this gene in mice also seems to confer greater resistance to certain cancers. Mutations in the p66shc gene in mice also increases longevity by up to 30%. One other effect of this mutation is to increase resistance to oxidative damage, another mechanism implicated in aging. While the effects of altering single genes have been studied for some time, new techniques such as DNA microarrays permit the simultaneous examination of gene expression in hundreds, even thousands of genes. What has emerged from these studies is the consistent pattern that long-lived individuals, including humans, show greater stability of gene expression across their adult life spans than younger or shorter-lived individuals. However, one problem with extrapolating from studies of simpler organisms is their biological distance from humans. Even though they may share similar genes and biochemical pathways, the effects are not always the same in humans as in the simpler models. Some mechanisms of aging do appear to be similar across different species. For instance, the SIR2 gene has been demonstrated as an important factor in S. cerevisiae longevity and its analog Sir-2.1 has a similar effect in C. elegans, but findings in Drosophila, considered by many scientists to be a more reliable model of human biology and genetics, are contradictory regarding the role of Sir2 in aging in this species.
 genes of the immune system, apolipoprotein E (APOE), angiotensin-converting enzyme (ACE), plasminogen activating inhibitor 1 (PAI-1), and p53, are associated with living past age ninety. Forms of several of these genes, such as APOE, ACE, and p53, are associated with increased risk of developing Alzheimer s disease, cardiovascular disease, and cancer, respectively. The association of these genes with longevity may be due to these disease associations, or it may be due to their direct influence on extending the human life span. Regardless, genes clearly influence aging and longevity, whether it is by influencing the development of life span-shortening diseases, or by positively influencing longevity independently of causing disease. The receptors include those that are targeted by drugs used to treat high cholesterol and triglyceride levels. Mark Mattson et al. One theory of aging assumes that the life span of a cell or organism is genetically determined—that the genes of an animal contain a program that determines its life span just as eye color is determined genetically, but what are the genes responsible Nobody knows,… Error theory: Kiecolt-Glaser, 2003; Ly et al., 2000) Stress resistance genes: In model systems such as C Elegans, extended longevity is often associated with increased stress resistance (see eg. Longo, 1999). Manipulations in C elegans that extend longevity have a strong correlation to stress resistance, in the form of oxidative stress or inflammation probes. Cell lines from long lived species are stress resistant. Stress resistans in vitro also correlates with mamalian longevity (Kapahi et al., 1999). Cells taken from individuals with progeroid syndromes are more susceptible to stress (Gebhart et al., 1988). Genetic Expression - In certain instances, individual genes have been found that appear to directly affect longevity or rate of aging, but they do so through different mechanisms. Over expression of the urokinase plasminogen activator (uPA) in mice brains results in a 20% increase in longevity, probably by producing effects similar to Caloric Restriction. Urokinase plasminogen-activator is a serine protease that is important in embryonic development, aging and the onset of pathogenic conditions. Interestingly enough, deletion of this gene in mice also seems to confer greater resistance to certain cancers. Mutations in the p66shc gene in mice also increases longevity by up to 30%. One other effect of this mutation is to increase resistance to oxidative damage, another mechanism implicated in aging. While the effects of altering single genes have been studied for some time, new techniques such as DNA microarrays permit the simultaneous examination of gene expression in hundreds, even thousands of genes. What has emerged from these studies is the consistent pattern that long-lived individuals, including humans, show greater stability of gene expression across their adult life spans than younger or shorter-lived individuals. However, one problem with extrapolating from studies of simpler organisms is their biological distance from humans. Even though they may share similar genes and biochemical pathways, the effects are not always the same in humans as in the simpler models. Some mechanisms of aging do appear to be similar across different species. For instance, the SIR2 gene has been demonstrated as an important factor in S. cerevisiae longevity and its analog Sir-2.1 has a similar effect in C. elegans, but findings in Drosophila, considered by many scientists to be a more reliable model of human biology and genetics, are contradictory regarding the role of Sir2 in aging in this species.")
61
Many genes shown to influence life span are involved in DNA damage repair and protection.

62
Progeria: Two forms in humans; Werner's syndrome (adult-onset progeria) and Hutchinson-Gilford syndrome (juvenile-onset progeria). Most clinicians believe that progeria is segmental aging Mutation in Werner’s codes for a DNA helicase (DNA repair/unwind) From: Accelerated Aging: Progeria Human progeria comes in two major forms, Werner's syndrome (adult-onset progeria) and Hutchinson-Gilford syndrome (juvenile-onset progeria). Werner's patients are usually diagnosed in early maturity and have an average life span of forty-seven years. Hutchinson-Gilford patients are usually diagnosed within the first two years of life and have an average life span of thirteen years. The latter syndrome is often simply termed "progeria" and both are sometimes lumped together as progeroid syndromes. Progeria's Effects There is considerable controversy as to whether or not progeria is a form of aging at all. Most clinicians believe that progeria is truly a form of early aging, although only a segmental form in which only certain specific tissues and cell types of the body age early. Hutchinson-Gilford children show what appears to be early aging of their skin, bones, joints, and cardiovascular system, but not of their immune or central nervous systems. Clinical problems parallel this observation: They suffer from thin skin and poor skin healing, osteoporosis, arthritis, and heart disease, but do not have more infections than normal children and they do not have early dementia. Death is usually due to cardiovascular disease, especially heart attacks and strokes, yet Hutchinson-Gilford children lack normal risk factors associated with these diseases, such as smoking, high cholesterol, hypertension, or diabetes. Clinically, the children appear old, with thin skin, baldness, swollen joints, and short stature. They do not go through puberty. The face is strikingly old in appearance. The typical Hutchinson-Gilford child looks more like a centenarian than like other children, and may look more like other progeric children than like members of their own families. There is no effective clinical intervention. Inheritance of Progeria The segmental nature of progeria is perhaps its most fascinating feature. If progeria is actually a form of aging gone awry, then this implies that aging is more than merely wear and tear on the organism. If progeria is a genetically mediated, segmental form of aging, this may imply that aging itself is genetically mediated and, like other genetic disease, is not only the outcome of genetic error but might be open to clinical intervention. Supporting this observation, there are a number of other less well-known forms of progeria, including acrogeria, metageria, and acrometageria, as well as several dozen human clinical syndromes and diseases with features that have been considered to have progeroid aspects. The latter category includes Wiedemann-Rautenstrauch, Donohue's, Cockayne's, Klinefelter's, Seip's, Rothmund's, Bloom's, and Turner's syndromes, ataxia telangiectasia, cervical lipodysplasia, myotonic dystrophy, dyskeratosis congenita, and trisomy 21 (Down syndrome). In each of these cases, there are features that are genetic and that have been considered segmental forms of aging. In the most well-known of these, trisomy 21, the immune and central nervous systems both appear to senesce early, in contrast with Hutchinson-Gilford progeria, in which the opposite occurs. Bolstering the suggestion that this is a form of segmental progeria, trisomy 21 patients are prone to both infections and early onset of a form of Alzheimer's dementia. The gene that is mutated in Werner's syndrome is known to code for a DNA helicase. This enzyme unwinds DNA for replication, transcription, recombination, and repair. The inability to repair DNA may explain the features of premature aging, as well as the increased rate of cancer in Werner's syndrome patients. Another mutated helicase is responsible for Bloom's syndrome. Both conditions are inherited as autosomal recessive disorders. Data suggesting that Hutchinson-Gilford progeria is genetic is circumstantial. The disease is presumptively caused by a sporadic (one in eight million live births), autosomal dominant mutation, although a rare autosomal recessive mutation is not impossible. The helicase abnormality that causes Werner's syndrome is not present in Hutchinson-Gilford cells. There is a slight correlation with the paternal age at conception. Whatever the mechanism, it appears to operate prior to birth; several neonatal cases have been reported. Germinal Mosaicism Cellular data, particularly regarding structures called telomeres, suggests that some of the cells from Hutchinson-Gilford patients are prone to early cell senescence. Telomeres are special DNA structures at the tips of the chromosomes. These telomeres gradually shorten over time, and this shortening is associated with some aspects of cellular aging. Skin fibroblasts from Hutchinson-Gilford patients have shorter than normal telomeres and consequently undergo early cell senescence. At birth, the mean telomere length of these children is equivalent to that of a normal eighty-five-year-old. Introduction of human telomerase into such cells leads to reextension of the telomeres and results in normal immortalization of these progeric cell cultures. Clinical interventional studies using this strategy in humans are pending. Predictably, circulating lymphocytes of Hutchinson-Gilford children have normal telomere lengths, in keeping with their normal immune function. Research thus far suggests that progeria may not be so much a genetic disease as it is an "epigenetic mosaic disease." In progeria, this means that the genes are normal, but the abnormally short telomere length in only certain cells lines causes an abnormal pattern of gene expression. The senescent pattern of gene expression in specific tissues results in the observed clinical disease of progeria. Although consistent with all known laboratory and clinical data, the actual genetic mechanisms that underlie Hutchinson-Gilford progeria are still uncertain and arguable (the gene for Werner's syndrome, however, has been cloned). The question of what causes progeria holds a fascination largely for what it may tell us about the course of aging itself. From:
 From: Accelerated Aging: Progeria. Human progeria comes in two major forms, Werner s syndrome (adult-onset progeria) and Hutchinson-Gilford syndrome (juvenile-onset progeria). Werner s patients are usually diagnosed in early maturity and have an average life span of forty-seven years. Hutchinson-Gilford patients are usually diagnosed within the first two years of life and have an average life span of thirteen years. The latter syndrome is often simply termed progeria and both are sometimes lumped together as progeroid syndromes. Progeria s Effects. There is considerable controversy as to whether or not progeria is a form of aging at all. Most clinicians believe that progeria is truly a form of early aging, although only a segmental form in which only certain specific tissues and cell types of the body age early. Hutchinson-Gilford children show what appears to be early aging of their skin, bones, joints, and cardiovascular system, but not of their immune or central nervous systems. Clinical problems parallel this observation: They suffer from thin skin and poor skin healing, osteoporosis, arthritis, and heart disease, but do not have more infections than normal children and they do not have early dementia. Death is usually due to cardiovascular disease, especially heart attacks and strokes, yet Hutchinson-Gilford children lack normal risk factors associated with these diseases, such as smoking, high cholesterol, hypertension, or diabetes. Clinically, the children appear old, with thin skin, baldness, swollen joints, and short stature. They do not go through puberty. The face is strikingly old in appearance. The typical Hutchinson-Gilford child looks more like a centenarian than like other children, and may look more like other progeric children than like members of their own families. There is no effective clinical intervention. Inheritance of Progeria. The segmental nature of progeria is perhaps its most fascinating feature. If progeria is actually a form of aging gone awry, then this implies that aging is more than merely wear and tear on the organism. If progeria is a genetically mediated, segmental form of aging, this may imply that aging itself is genetically mediated and, like other genetic disease, is not only the outcome of genetic error but might be open to clinical intervention. Supporting this observation, there are a number of other less well-known forms of progeria, including acrogeria, metageria, and acrometageria, as well as several dozen human clinical syndromes and diseases with features that have been considered to have progeroid aspects. The latter category includes Wiedemann-Rautenstrauch, Donohue s, Cockayne s, Klinefelter s, Seip s, Rothmund s, Bloom s, and Turner s syndromes, ataxia telangiectasia, cervical lipodysplasia, myotonic dystrophy, dyskeratosis congenita, and trisomy 21 (Down syndrome). In each of these cases, there are features that are genetic and that have been considered segmental forms of aging. In the most well-known of these, trisomy 21, the immune and central nervous systems both appear to senesce early, in contrast with Hutchinson-Gilford progeria, in which the opposite occurs. Bolstering the suggestion that this is a form of segmental progeria, trisomy 21 patients are prone to both infections and early onset of a form of Alzheimer s dementia. The gene that is mutated in Werner s syndrome is known to code for a DNA helicase. This enzyme unwinds DNA for replication, transcription, recombination, and repair. The inability to repair DNA may explain the features of premature aging, as well as the increased rate of cancer in Werner s syndrome patients. Another mutated helicase is responsible for Bloom s syndrome. Both conditions are inherited as autosomal recessive disorders. Data suggesting that Hutchinson-Gilford progeria is genetic is circumstantial. The disease is presumptively caused by a sporadic (one in eight million live births), autosomal dominant mutation, although a rare autosomal recessive mutation is not impossible. The helicase abnormality that causes Werner s syndrome is not present in Hutchinson-Gilford cells. There is a slight correlation with the paternal age at conception. Whatever the mechanism, it appears to operate prior to birth; several neonatal cases have been reported. Germinal Mosaicism. Cellular data, particularly regarding structures called telomeres, suggests that some of the cells from Hutchinson-Gilford patients are prone to early cell senescence. Telomeres are special DNA structures at the tips of the chromosomes. These telomeres gradually shorten over time, and this shortening is associated with some aspects of cellular aging. Skin fibroblasts from Hutchinson-Gilford patients have shorter than normal telomeres and consequently undergo early cell senescence. At birth, the mean telomere length of these children is equivalent to that of a normal eighty-five-year-old. Introduction of human telomerase into such cells leads to reextension of the telomeres and results in normal immortalization of these progeric cell cultures. Clinical interventional studies using this strategy in humans are pending. Predictably, circulating lymphocytes of Hutchinson-Gilford children have normal telomere lengths, in keeping with their normal immune function. Research thus far suggests that progeria may not be so much a genetic disease as it is an epigenetic mosaic disease. In progeria, this means that the genes are normal, but the abnormally short telomere length in only certain cells lines causes an abnormal pattern of gene expression. The senescent pattern of gene expression in specific tissues results in the observed clinical disease of progeria. Although consistent with all known laboratory and clinical data, the actual genetic mechanisms that underlie Hutchinson-Gilford progeria are still uncertain and arguable (the gene for Werner s syndrome, however, has been cloned). The question of what causes progeria holds a fascination largely for what it may tell us about the course of aging itself. From:")
63
Progeria Werner’s syndrome: Hutchinson-Gilford:
- chromosome instability syndromes Inability to repair DNA Increased rate of cancer mutated helicase inherited as autosomal recessive Hutchinson-Gilford: no helicase abnormality The pattern of inheritance is uncertain have shorter than normal telomeres undergo early cell senescence.

64
Mitochondrial Aging Mitochondrial DNA is extra sensitive to damage, such as oxidative stress because it does not have repair mechanisms like normal DNA

65
Summary of mitochondrial theory:
Decreased activity of electron transport chain complex with aging Increased release of ROS Alterations in mitochondrial apoptosis pathways (Bax/Bcl-2 etc) Lack of repair mechanisms mtDNA Slower mitochondrial turnover accumulates mtDNA mutations the faster the mitochondria turn over, the better – it’s a good idea to replace mitochondrial populations before too much mtDNA damage accumulates. All these factors together suggest that the mitochondria hold important clues to what happens with aging. They are more sensitive than other DNA, repair mechanisms are less efficient and they partake in some of the most important processes, such as energy production and apoptotic pathways (Refs: Remmen et al., Exp. Gerontol. (2001) 36:957-68; Sastre et al., ANN NY Acad Sci 959: , Sastre et al: Females may live longer than males because the oxidant production by mt is much lower in females than males. However, OVX removes that advantage, and antiox treatment can increase it.
 Lack of repair mechanisms mtDNA. Slower mitochondrial turnover accumulates mtDNA mutations. the faster the mitochondria turn over, the better – it’s a good idea to replace mitochondrial populations before too much mtDNA damage accumulates. All these factors together suggest that the mitochondria hold important clues to what happens with aging. They are more sensitive than other DNA, repair mechanisms are less efficient and they partake in some of the most important processes, such as energy production and apoptotic pathways (Refs: Remmen et al., Exp. Gerontol. (2001) 36:957-68; Sastre et al., ANN NY Acad Sci 959: , Sastre et al: Females may live longer than males because the oxidant production by mt is much lower in females than males. However, OVX removes that advantage, and antiox treatment can increase it.")
66
Genomic Alterations with Aging:
Intact telomeric DNA Intact nuclear DNA Intact mtDNA Endogenous oxidative stress DNA repair Damaged/shorter telomeric DNA Damaged nuclear DNA Damaged mtDNA To summarize, Aging overall involves alterations in both telomeric DNA, global DNA, and mitochondrial DNA. The continued mitosis gives rise to increased risk for mutations, leading to malfunction of proteins and /or cancer development. Cell cycle arrest senescence Apoptosis Cell loss Mutation and cancer Diminished energy production Aging

Similar presentations