Download presentation
Presentation is loading. Please wait.

1
By Prof. MANAL ELDEFRAWY Awareness of some IEM 2011

2
Objectives When to suspect a metabolic disorder? Immediate investigations Specific investigations Which emergency measures? Diagnostic algorithm Case presentation

3
Single gene defect in an enzyme or transport protein, which results in a block in a metabolic pathway of proteins,CHO, fats, or complex molecules. Effects are due to: toxic accumulations of substrates before the block, intermediates from alternative metabolic pathways, defects in energy production caused by a deficiency of products beyond the block, or a combination of these metabolic deviations

4
The incidence, collectively, is estimated to be approximately 1 in 4000 live births The inheritance is mostly autosomal recessive with male to female ratio of 1:1

5
When To think Metabolic Time & Pattern of Onset Neonatal period A) Intoxication type Typically born healthy, symptom free period Deterioration with poor feeding, vomiting, lethargy, apnea seizures, coma. e.g. a.acidopathies, Organic acidemia, UCDs, CHO intolerance. B) Energy deficiencies No apparent Symptom free Overwhelming neurologic deterioration apnea, seizures, coma e.g. FAO defects, Mitochondrial disorders, peroxisomal disorders
 Energy deficiencies No apparent Symptom free Overwhelming neurologic deterioration apnea, seizures, coma e.g. FAO defects, Mitochondrial disorders, peroxisomal disorders.")
6
When To Think Metabolic? Beyond neonatal period Lethargy or just not doing well Refusal to feed poor suckling Vomiting Poor weight gain Hepatomegaly Tachypnea Hypothermia Axial hypotonia Limb hypotonia Abnormal movements(pedalling, tremors) Altered conscious, seizures, coma Multivisceral failure
 Altered conscious, seizures, coma Multivisceral failure.")
7
When to think metabolic? Additional factors Consanguinity Unexplained Neonatal deaths Unexplained Deterioration with symptomatic treatment

8
Initial investigations parallel to Sepsis screening Urine Ketonuria bedside test Unusual Odour Unusual colour Reducing subtances Blood Glucose Electrolytes: Na,K,cl,Ca Ammonia Blood gases Anion gap Transaminases Prothrombin T&C & INR Urea & creatinine Uric acid Lactic acid CK CBC

9
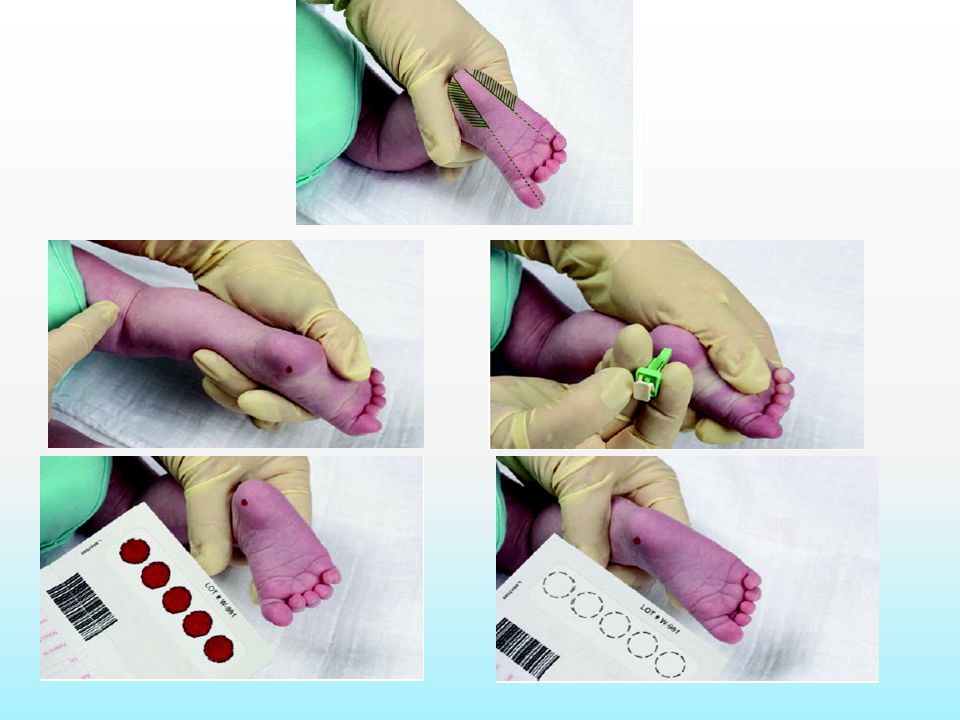
Second Line Evaluation Urine and plasma aminogram Urine organic acid profile by GC-MS Plasma carnitine and Acyl carnitine profile: Increase carnitine ester in fatty acid oxidation defect, organic academias, ketosis Dried blood spot analyzed by tandem mass spectrometry (MS/MS).
.")
11
Second Line Evaluation(cont.) CSF anaylsis NKH is diagnosed by the presence of elevated CSF to plasma glycine ratio Decrease CSF glucose/to blood glucose…..glucose transporter defect Peroxisomal function Test Plasma very long chain fatty acids (VLCFAS), Phytanic acid, erythrocyte plasmalogen levels Stored samples Frozen whole and heparinized bl., serum, CSF & urine (store at -20°C).
 CSF anaylsis NKH is diagnosed by the presence of elevated CSF to plasma glycine ratio Decrease CSF glucose/to blood glucose…..glucose transporter defect Peroxisomal function Test Plasma very long chain fatty acids (VLCFAS), Phytanic acid, erythrocyte plasmalogen levels Stored samples Frozen whole and heparinized bl., serum, CSF & urine (store at -20°C).")
12
Emergent treatment of IEMs (acute life-threatening ) Establish airway, breathing, circulation. Avoid lactated Ringer’s. Avoid hypotonic fluid load due to the risk of cerebral edema (if hyperammonemia is present). NPO :Discontinue intake of offending agents; (especially no protein, galactose, or fructose) Provide adequate glucose to prevent catabolism. (Fluid boluses D10 normal saline).
. NPO :Discontinue intake of offending agents; (especially no protein, galactose, or fructose) Provide adequate glucose to prevent catabolism. (Fluid boluses D10 normal saline)..")
13
Most IEMs with acute life-threatening presentation can be categorized based on finding of at least 1 of the following : Hyperammonemia Metabolic acidosis Hypoglycemia Jaundice and Liver dysfunction

14
Hypoglycemia Severe, persistent, without other etiology If with metabolic acidosis e.g. OA, GSD Type 1, Fructose 1, 6 Diphosphatase deficiency, FAO (hallmark Non ketotic) N.B :Hypoglycemia (plasma glucose level < 50 mg/dL)
 N.B :Hypoglycemia (plasma glucose level < 50 mg/dL).")
15
Hypoglycemia in neonatal period

16
Treatment of Hypoglycemia D10 to D15 with electrolytes 8-12 mg/kg/min IV to maintain serum glucose leve at 120–170mg/dL which should prevent catabolism. High-volume maintenance fluid 1-1.5 times maintenance will also promote urinary excretion of some toxic metabolites. If necessary, treat hyperglycemia with insulin (0.2-0.3 IU/kg/h) to maintain glucose level in the desired range.
 to maintain glucose level in the desired range..")
17
Hyperammonemia Excess ammonia (about80% dietary and waste nitrogen) is converted to urea in the liver through urea cycle. The five enzymes that make up the urea cycle are regulated long term by the quantity of dietary protein. Ammonia level : >100 mcg/dL in the neonate > 80 mcg/dL beyond the neonatal period is considered elevated & toxic.

18
Hyperammonemia This can lead to Encephalopathy and death OR Devastating neurological sequelae. Neurological sequelae and survival depend on the length of hyperammonemic coma(better prognosis <36h or<2days).
..")
19
Hyperammonemia Significant hyperammonemia is observed in Urea cycle defect Organic acidemia Fatty acid oxidations defects THAN Ammonia is highest in the UCDs often exceeding 1000 mcg/dL and causing primary respiratory alkalosis sometimes with compensatory metabolic acidosis. Ammonia in OA, if elevated, rarely exceeds 500 mcg/dL, and in FAO is usually less than 250 mcg/dL.

20
Hyperammonemi a Normal PH or alkalosis Acidosis Normoglycemi a Hypoglycemia Hypoketosis Fatty acid oxidation (FAO) Organic acidemia Pyruvate metabolism Plasma aminoacids Urea cycle disorders Increased citrulline ASA present Low/Normal citrulline Increased Citrulline ASA absent Urine Orotic acid Citrullinemia OTC deficiency CPS deficiency THAN Argininosuccinic acidemia
 Organic acidemia Pyruvate metabolism Plasma aminoacids Urea cycle disorders Increased citrulline ASA present Low/Normal citrulline Increased Citrulline ASA absent Urine Orotic acid Citrullinemia OTC deficiency CPS deficiency THAN Argininosuccinic acidemia")
21
The therapeutic protocol include : Avoidance of nitrogen intake by nasogastric infusion of a protein-free formula. Adequate caloric intake (80-120 kcal/kg/d) by glucose (10 – 20 g/kg ). Sodium benzoate (250-500 mg/kg/d),more recently phenylbutyrate (250mg/kg/d) peroral IV administration of arginine (250-500mg/kg/d) Carnitine (250-500 kg/d) Hydroxycobalamin ( 1mg/d) Biotin( 10 mg/d) Insulin may be added to utilize its anabolic effect (0.1 -1 IU /kg/ hr if bl. glucose > 200 mg/dl ) Dialysis in patients not responding to pharmacological therapy
 by glucose (10 – 20 g/kg ). Sodium benzoate ( mg/kg/d),more recently phenylbutyrate (250mg/kg/d) peroral IV administration of arginine ( mg/kg/d) Carnitine ( kg/d) Hydroxycobalamin ( 1mg/d) Biotin( 10 mg/d) Insulin may be added to utilize its anabolic effect ( IU /kg/ hr if bl. glucose > 200 mg/dl ) Dialysis in patients not responding to pharmacological therapy.")
22
Case presentation A 3 years old boy presented to ER with disturbed level of consciousness of 2 days duration triggered by fever and associated with one brief attack of generalized tonic clonic seizures. The day before he ate high protein containing food which was followed by : period of hyperirritability and profuse vomiting then gradual and progressive lapse in the level of consciousness which worsen upon the introduction of depakene to control the seizure.

23
Past history : revealed two similar episodes of disturbed conscious level : -The first was at 7days in neonatal period. CT at that time revealed brain edema. -The second episode was at the age of 18 months. Examination : revealed a semi conscious child with generalized axial and limb hypotonia, RR 50/m.

24
Lab investigation Blood gases: ph=7.5, Pco2=24,Hco3=18 Ammonia level =250mg/dl Lactate = 3.2 mmol/l ( n < 2.1 ) Normal LFT &KFT CBC: mild neutrophilia Salient features Disturbed conscious level respiratory alkalosis Hyperammonemia
 Normal LFT &KFT CBC: mild neutrophilia Salient features Disturbed conscious level respiratory alkalosis Hyperammonemia")
25
MS/MS revealed very high level of citrulline Orotic acid in urine was increased Final diagnosis: citrullinemia

27
MS/MS Normal Citrullinemi a

28
HHH syndrome One year & 1mth old boy with severe failure to thrive, excessive sleepiness. The condition started at the age of 4mths after feeding baby with yogurt. He started to have recurrent attacks of vomiting sometimes with diarrhea,admitted to hospital for IV fluids. On examination: Fair complexion, apathy, GDD (motor &mental). Normal abd. Exam. Pt. weighted 6 kg, H.C. 42 cm, length 69 cm (all< 3 rd percentile for age)
. Normal abd. Exam. Pt. weighted 6 kg, H.C. 42 cm, length 69 cm (all< 3 rd percentile for age).")
29
HHH syndrome

30
HHH cont. Investigations: HGB : 10.5, normocytic,normochromic a. ABG : metabolic alkalosis ( repeatedly) Ammonia : high ( repeatedly) > 300ug/dl TMS: ( HHH syndrome) Hyperammonemia, Hyperornithinemia and Homocitrullinemia
 Ammonia : high ( repeatedly) > 300ug/dl TMS: ( HHH syndrome) Hyperammonemia, Hyperornithinemia and Homocitrullinemia.")
31
HHH syndrome after therapy Treat. hyperammonemia ornithine supplementation ????

32
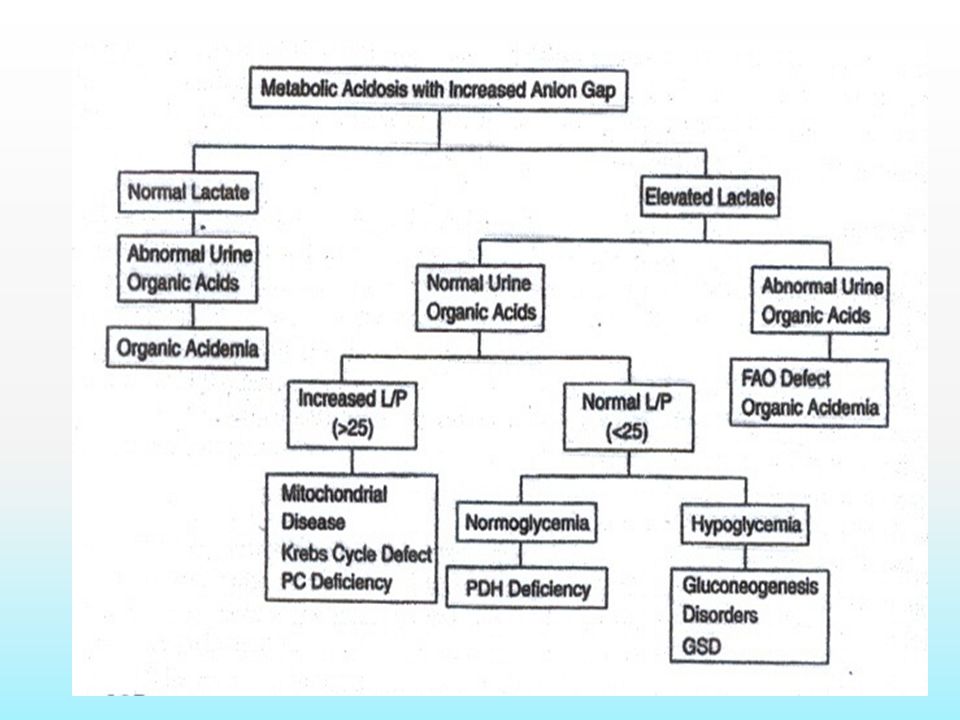
Metabolic Acidosis Among the inborn errors, the largest group typically associated with overwhelming metabolic acidosis in infancy is the group of organic acidemias Methylmalonic acidemia, Propionic acidemia, and Isovaleric acidemia.

33
Important Laboratory Findings in Organic Acidemia Hyperlactic acidemia Neutropenia and thrombocytopenia Hyperammonemia Ketosis

35
Emergency management of metabolic Acidosis Metronidazole(10-20mg/kg) and neomycin (50mg/kg) reduce the level of priopionic acid and methyl malonic acid in body fluid Antibiotic as clindamycin and vancomycin. Hyperammonemic episodes should be treated promptly with Na benzoate(250mg/kg in10% glucose) Aggressive fluid and electrolyte therapy is essential in acute ketoacidotic crisis: 150mg-200mg/kg of 10% glucose and isotonic NAHCO3 until acidosis is corrected : 0.25-0.5 mEq/kg/h (up to 1-2 mEq/kg/h) IV Consider hemodialysis ; if intractable acidosis, (peritoneal dialysis, hemofiltration, exchange transfusion much less effective).
 Aggressive fluid and electrolyte therapy is essential in acute ketoacidotic crisis: 150mg-200mg/kg of 10% glucose and isotonic NAHCO3 until acidosis is corrected : mEq/kg/h (up to 1-2 mEq/kg/h) IV Consider hemodialysis ; if intractable acidosis, (peritoneal dialysis, hemofiltration, exchange transfusion much less effective)..")
36
Methylmalonic acidemia Marked failure to thrive severe metabolic acidosis ketosis Severe psychomotor retardation Encephalopathy Dystonia Recurrent seizures Peritoneal dialysis

37
Brain MRI T2w MRI demonstrating bilateral and symmetrical high signal intensity lesions in the globus pallidus

38
MMA Date : 9/2010 - 2 years old male -3mths ago he developed recurrent attacks of G.E. & M.A. -Then he suffered from coma for nearly a week ?. -After that he developed severe failure to thrive. Expanded metabolic screen using LC-MS/MS showed MMA

39
Ethyl Malonic Acidemia A.A, the 3 rd child of first degree consanguineous parents whose birth date is 8/2008 The patient presented at the age of 9 months with GDD and intractable seizures Antenatal, natal and perinatal period were uneventfull Clinical exam revealed, microcephaly, microphthalmia, frontal bossing, hypertolerism, epicanthic folds, long filtrum and bat ears

40
Ethyl Malonic Acidemia Neurological exam revealed hypertonia, hyperreflexia, positive babinski, truncal hypotonia. Recurrent ecchymotic patches on the lower limb No episodes of metabolic decompensation or metabolic acidosis Pt. has a normal 4 years aged sister and mother had one abortion

41
Ethyl Malonic Acidemia TMS result: elevated butyryl carnitine (C4) =4. micomol/L cut off value =1.5 micromol/L Urine organic acid analysis by GC-MS showed highly elevated Ethylmalonic acid and moderately elevated methylsuccinic acid no dicarboxylic aciduria and no lactic aciduria

42
Brain MRI in EMA T2w Brain MRI showing widening of extraaxial CSF spaces, and sylvian fissure, picture of severe brain atrophy TW1 Brain MRI showing frontotemporal atrophy

43
Ethyl malonic acidemia Five mths old girl coming from Gaza(during 25 January revolution 2011) with recurrent attacks of diarrhea, vomiting,with severe napkin dermatitis and failure to thrive. Clinical exam. revealed, microcephaly, frontal bossing, hypertolerism, epicanthic folds, long filtrum and bat ears She weighted 3 kg, she had hyperammonemia, met. acidosis. TMS : Ethyl malonic acidemia

44
Pyroglutamic aciduria 3 mths old boy,1 st baby Admitted to PICU due to DCL, shortness of breath, increased yellowish discoloration of skin after severe attacks of vomiting for previous 2 days not associated by diarrhea.

45
History The condition started at the age of 5 days by Jaundice which was diagnosed as physiological, no treatment. For the following 3 month the mother noticed gradual abd. distention and no improvement in colour. On examination the baby was lethargic with tachypnea, hepatomegaly (span 10cm ) and cholestatic jaundice ( T=9.6, d=5.2). He was ventilated but it was difficult to be weaned from vent. as he developed cardiac problems as severe bradycardia and arrest.
 and cholestatic jaundice ( T=9.6, d=5.2). He was ventilated but it was difficult to be weaned from vent. as he developed cardiac problems as severe bradycardia and arrest..")
46
Investigations CBC: HB 10gm%, WBC :24,000 Bil total: 9.6 Direct :5.2 mg/dl ALT :115. PT :26, INR:2.9 Urea :30, creatinine:0.7 CRP: 12 ABG : variable with increased anion gap 59 Bl. Ammonia : 607---- 230---303 ug/dl MS/MS : Pyroglutamic aciduria Urine organic acids was done

47
The baby was treated with Na benzoate and glutathione analogs, vit C and vit E. He started to gain consciousness and he was weaned from the vent. The jaundice and hepatomegaly decreases.

48
Pyroglutamic aciduria after therapy

49
2.8 years old At the age of 2y, she developed recurrent attacks of convulsions and was treated with antiepileptic drugs At 2.1 y, she suffered recurrent attacks of RD with metabolic acidosis, no hypogycemia,no hep. Then she went in coma for 1 d. Bl Ammonia level was elevated so she received Na benzoate She improved for 2w then amm. increased again. MS/MS was normal ALT,AST were 2-3 times normal There was no hypoglycemia during the illness Liver span 10 cm, firm Liver biopsy 10/8/2010 metabolic LD ---- GSD

51
Hepatocellular necrosis, acute or subacute GALACTOSEMIA There are three known enzymatic errors. The most common defect is decreased activity of galactose 1-phosphate uridyltransferase (GALT) Clinical manifestations include : lethargy, hypotonia, jaundice. Hypoglycemia, elevated liver enzymes Coagulopathy
 Clinical manifestations include : lethargy, hypotonia, jaundice. Hypoglycemia, elevated liver enzymes Coagulopathy.")
52
Diagnosis Reducing substances in infants urine with simultaneous normal or low blood sugar while the infant is being fed breast milk or a formula containing lactose.low blood sugar enzyme activity measurement in erythrocytes. enzyme Prenatal diagnosis by direct measurement of the enzyme activity(GALT). Prenatal diagnosis
. Prenatal diagnosis.")
53
GALACTOSEMIA

54
Treatment The main is lactose-free formula followed by dietary restriction of all lactose-containing foods later in life. Untreated infants may have severe growth failure, mental retardation, cataracts, ovarian failure, and liver cirrhosis. Despite early and adequate intervention, some children still may develop milder signs of these clinical manifestations.

56
Hereditary fructose intolerance (HFI) Alternative names Fructosemia; Fructose intolerance; Fructose aldolase B- deficiency; Fructose 1, 6 bisphosphate aldolase deficiency An autosomal recessive disorder Although glucose may still be released through the breakdown of glycogen,it cannot be synthesized from gluconeogenesis, resulting in severe hypoglycaemiaglycogengluconeogenesishypoglycaemia
 Alternative names Fructosemia; Fructose intolerance; Fructose aldolase B- deficiency; Fructose 1, 6 bisphosphate aldolase deficiency An autosomal recessive disorder Although glucose may still be released through the breakdown of glycogen,it cannot be synthesized from gluconeogenesis, resulting in severe hypoglycaemiaglycogengluconeogenesishypoglycaemia")
57
Hereditary fructose intolerance (HFI)
")
58
Tests that confirm the diagnosis Positive urine test for reducing substances. Hypoglycemia, especially after receiving fructose/sucrose. Abnormally high amounts of amino acids and salts in urine Enzyme studies Treatment Complete elimination of fructose and sucrose from the diet is an effective treatment for most patients.

59
TYROSINEMIA TYPE I (TTI) Autosomal recessive disorder Incidence of 1:100,000 to 1:120,000. The defect is in fumaryl acetoacetate hydrolase, resulting in accumulation of metabolites such as fumarylacetoacetate and malelylacetoacetate which are alkylating agents that cause damage to DNA and predisposition to HCC. One of the byproducts of these metabolites is succinyl acetone, which is a diagnostic marker for tyrosinemia.

60
Presentation of TTI Include : Acute liver failure, chronic liver disease, HCC. Renal tubular dysfunction. Episodic porphyria-like neurological episodes(42% of patients )caused by succinyl acetone inhibiting the metabolism of aminolevulinic acid. Neurological crises began at a mean age of 1 year. Episodes included severe pain with extensor hypertonia, vomiting or paralytic ileus, muscle weakness, and self- mutilation.
caused by succinyl acetone inhibiting the metabolism of aminolevulinic acid. Neurological crises began at a mean age of 1 year. Episodes included severe pain with extensor hypertonia, vomiting or paralytic ileus, muscle weakness, and self- mutilation..")
61
TYROSINEMIA TYPE I (TTI)
")
62
Diagnosis Serum amino acid patterns may exhibit high levels of tyrosine, phenylalanine and methionine. Generalized aminoaciduria. High levels of alpha-fetoprotein are observed. succinyl acetone in urine and serum is a diagnostic marker for tyrosinemia. Enzyme measurement in lymphocytes &RBC

63
Product ion scans of derivatized SA (top) and 5,7- dioxooctanoic acid (internal standard; bottom) and fragmentation of SA-hydrazone (inset). A simple method for quantifying SA in dried blood spots has been described by Allard et al.,2004. Inclusion of SA in our existing screening program could be achieved with little additional manual work.

64
The medical management of TTI Has changed considerably with the introduction of 2-(2-nitro-4-trifluoromethylbenzol)-1,3- cyclohexendiome (NTBC) in 1992. NTBC blocks the second step in tyrosine degradation, thus preventing formation of the alkylating metabolites. Currently, the indication for transplantation includes treatment failure or development of HCC.

65
WILSON'S DISEASE An autosomal recessive disease. The specific molecular defect resides within a copper-transporting ATPase encoded by a gene on chromosome 13. Affected patients exhibit impaired biliary excretion of copper; this leads to copper accumulation in the liver, brain, cornea, and kidneys.

66
Clinical presentation The most common presentation is that of postnecrotic cirrhosis with hepatic dysfunction and portal hypertension or chronic active hepatitis. The disease may manifests as fulminant hepatic failure; with massive liver necrosis, coincident hemolysis.

67
Diagnosis A useful screening test is low serum ceruloplasmin. It is < 20 mg per dl in 85% of patients. The presence of Kayser-Fleischer rings. Increased urinary excretion of copper. Elevated liver copper on biopsy. Patients with > 250 µg copper per gram of dry liver tissue are considered to have Wilson's disease. Measurement of urinary copper excretion in response to oral penicillamine challenge.

68
The Kayser–Fleischer ring around the periphery of the cornea caused by deposition of copper in Descemet's membrane

69
Hyperintensities in the bilateral basal ganglia and thalami shown by T2-weighted MRI of the brain Das SK and Ray K (2006) Wilson's disease: an update Nat Clin Pract Neurol 2: 482–493
 Wilson s disease: an update Nat Clin Pract Neurol 2: 482–493")
70
Treatment Copper chelation improves survival. Treatment includes D-penicillamine, triethylene tetramine dihydrochloride (tientene; generally used in D- penicillamine-intolerant patients), and oral zinc. Once initiated, therapy must be continued for life; discontinuation can result in rapid deterioration. liver transplantation in patients with fulminant hepatic failure or decompensated cirrhosis, provides effective therapy.
, and oral zinc. Once initiated, therapy must be continued for life; discontinuation can result in rapid deterioration. liver transplantation in patients with fulminant hepatic failure or decompensated cirrhosis, provides effective therapy..")
Similar presentations




>")











.>")



 IS A METABOLISM DISORDER PASSED DOWN THROUGH FAMILIES IN WHICH THE BODY CANNOT BREAK DOWN CERTAIN PARTS OF PROTEINS. URINE.>")

