Download presentation
Presentation is loading. Please wait.

1
General Pathology: Immunodeficiency and Transplantation
Lorne Holland, M.D.

2
Immunodeficiency A failure of the immune system to respond to usual stimuli Causes are varied, but can be grouped into defects of humoral immunity, cell-mediated immunity, phagocytosis and complement May be inherited (primary) or acquired (secondary)
 or acquired (secondary)")
3
Humoral Deficiencies Infancy
At birth, essentially all circulating immunoglobulins are maternal IgG As infant ages, maternal immunoglobulins are removed from circulation At the same time, baby is ramping up production of its own immunoglobulins (IgG, IgM, IgA) Early on, the decline in maternal concentrations happens a little faster than the infant can synthesize new immunoglobulins Around 3-6 months synthesis rates increase and total immunoglobulin concentrations begin to rise
 Early on, the decline in maternal concentrations happens a little faster than the infant can synthesize new immunoglobulins. Around 3-6 months synthesis rates increase and total immunoglobulin concentrations begin to rise.")
4
Humoral Deficiencies X-linked agammaglobulinemia (Bruton’s)
Failure of precurssor cells to differentiate into B-cells because of a lack of Bruton’s tyrosine kinase Responsible defective gene on X chromosome Initally present with recurrent pyogenic (bacterial) infections between 4 months and 2 years Replacement (transfusion) of pooled immunoglobulin can treat Increased risk of autoimmune diseases
 infections between 4 months and 2 years. Replacement (transfusion) of pooled immunoglobulin can treat. Increased risk of autoimmune diseases.")
5
Humoral Deficiency Hyper IgM syndrome
Normal or elevated levels of IgM, but no other immunoglobulins Most often, lack of CD40 ligand on T-cells leads to no isotype switching by B-cells and poor stimulation of macrophages CD40L is located on X chromosome so 70% of cases are seen in males Recurrent pyogenic infections and Pneumocystis pneumonia Treat with pooled immunoglobulin and prophylactic antibiotics or BMT

6
Humoral Deficiency Selective IgA deficiency
Normal or high immunglobulin concentrations expect IgA which is low or completely absent Typically clinically silent though those with no IgA may have increased rates of respiratory and GI infections Those with no IgA may also have anaphylactic reactions to IgA in blood products At higher risk for developing autoimmune diseases

7
Humoral Deficiency Common variable immunodeficiency
Low concentrations of one or more kind of immunoglobulin Usually much more modest increase in rate of pyogenic infections and so may not be diagnosed until adulthood Also at increased risk for a number of autoimmune diseases and lymphoid malignancies

8
Cell-mediated Deficiency
Severe combined immunodeficiency Impaired T-cell function which can also cause secondary impaired B-cell function Autosomal recessive form due to mutations in cytokine receptors which stimulate growth (25%) X-linked form due to mutation in adenosine deaminase necessary for DNA synthesis (50%) Present in the first weeks or months of life with recurrent infections of all kinds In absence of BMT (or gene therapy), life expectancy is typically 1-2 years
 X-linked form due to mutation in adenosine deaminase necessary for DNA synthesis (50%) Present in the first weeks or months of life with recurrent infections of all kinds. In absence of BMT (or gene therapy), life expectancy is typically 1-2 years.")
9
Cell-mediated Deficiency
Thymic hypoplasia (DiGeorge syndrome) Underdevelopment (absence) of the thymus leaves few places for T-cells to be “educated” Despite this, immunodeficiency is typically rather mild except in total absence Susceptible to fungal, viral, and protozoal infections (not typical bacteria species) Death can occurs due to associated abnormalities (e.g. cardiac malformations) Maybe prophylaxis with sulfamethoxazole/ trimethoprim (Bactrim, Septra) Transplantation of thymic tissue may be helpful
 Underdevelopment (absence) of the thymus leaves few places for T-cells to be educated Despite this, immunodeficiency is typically rather mild except in total absence. Susceptible to fungal, viral, and protozoal infections (not typical bacteria species) Death can occurs due to associated abnormalities (e.g. cardiac malformations) Maybe prophylaxis with sulfamethoxazole/ trimethoprim (Bactrim, Septra) Transplantation of thymic tissue may be helpful.")
10
Cell-mediated Deficiency
Wiscott-Aldrich syndrome Defects in both T-cells and B-cells along with thrombocytopenia and eczema Exact mechanism is unknown, but responsible gene is on the X chromosome and codes for proteins involved in cytoskeletal structure Untreated, death in early childhood BMT is only effective treatment Increased risk of hematologic malignancies

11
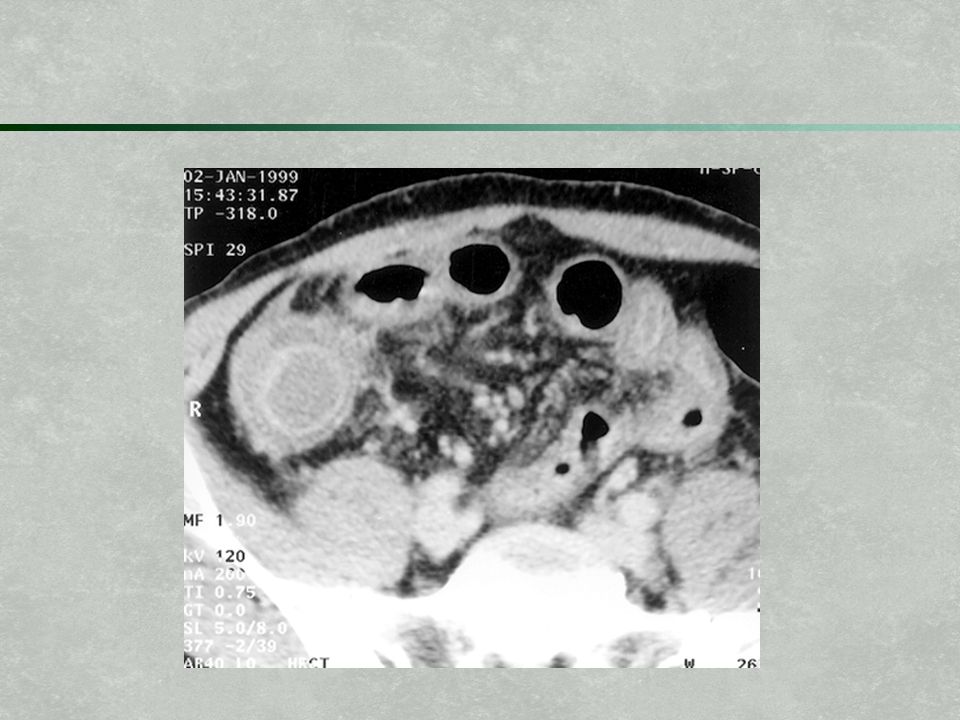
Phagocytosis Deficiency
Chronic granulomatous disease Failure of neutrophils to produce sufficient amounts of bacteriocidal products (reactive oxygen species) after phagocytosis Typically, presents as severe, recurrent skin infections by bacteria (staph) or fungus (candida, aspergillus) and/or pneumonia
 after phagocytosis. Typically, presents as severe, recurrent skin infections by bacteria (staph) or fungus (candida, aspergillus) and/or pneumonia.")
12
Phagocytosis Deficiency
Chronic granulomatous disease (cont) Ultimately, spreads systemically and form abscesses and granulomas in multiple organs (liver, bones) Maybe prophylaxis with sulfamethoxazole/ trimethoprim (Bactrim, Septra) Aggressive, early treatment of any potential infections Interferon therapy (may increase amounts of bacteriocidal products synthesized)
 Ultimately, spreads systemically and form abscesses and granulomas in multiple organs (liver, bones) Maybe prophylaxis with sulfamethoxazole/ trimethoprim (Bactrim, Septra) Aggressive, early treatment of any potential infections. Interferon therapy (may increase amounts of bacteriocidal products synthesized)")
13
Complement Deficiency

14
Complement Deficiency
C1, C4 and/or C2 present with autoimmune symptoms C3 presents with severe, recurrent bacterial infections (convergence of classic and alternate pathways as well as chemotaxis, opsonization) C5, C6, C6, C8 (MAC) presents with recurrent infections (meningitis, sepsis, arthritis) with encapsulated bacteria (Neisseria sp.)
 C5, C6, C6, C8 (MAC) presents with recurrent infections (meningitis, sepsis, arthritis) with encapsulated bacteria (Neisseria sp.)")
15
Secondary Immunodeficiency
Acquired Immunodeficiency Syndrome Caused by the human immunodeficency retrovirus (RNA) Two proteins on outside gp120 and gp41 Inside: p24, RNA, protease, reverse transcriptase, integrase Three key retroviral genes: gag, pol and env These gene products (proteins) must be cleaved by protease to become functional
 Two proteins on outside gp120 and gp41. Inside: p24, RNA, protease, reverse transcriptase, integrase. Three key retroviral genes: gag, pol and env. These gene products (proteins) must be cleaved by protease to become functional.")
16
HIV structure gag, pol env p24

17
Secondary Immunodeficiency
Acquired Immunodeficiency Syndrome Virus adheres to cells via interaction of gp120 and CD4 with help from chemokine receptors (CXCR4 and/or CCR5) T-cells, macrophages and other APCs are most vulnerable gp41 penetrates membrane and allows for transfer of viral RNA
 T-cells, macrophages and other APCs are most vulnerable. gp41 penetrates membrane and allows for transfer of viral RNA.")
18
Secondary Immunodeficiency
Acquired Immunodeficiency Syndrome Viral RNA reverse transcribed into DNA Viral DNA is inserted into host DNA by integrase Inserted DNA remains latent until infected cell is stimulated (by normal means) and begins to divide
 and begins to divide.")
20
Secondary Immunodeficiency
Acquired Immunodeficiency Syndrome Acute infection causes flu-like symptoms in some, but not all people Although destruction of CD4+ cells is occurring, symptoms may not appear for several years Time to seroconversion depends on testing method Typical anti-HIV antibody screens ~3 weeks HIV p24 antigen ~2 weeks HIV RNA ~1 week

21
Secondary Immunodeficiency
Acquired Immunodeficiency Syndrome After latent phase, clinical manifestations vary Generalized lymphadenopathy, diarrhea, night sweats, weight loss Meningitis, encephalopathy, neuropathy, dementia Unusual, opportunistic infections- Pneumocystis pneumonia, severe CMV or HSV, cerebral toxoplasmosis, unusual mycobactrial species, unusual fungal species, GI cryptosporidum Tumors- Kaposi’s sarcoma (HSV 8), non-Hodkins lymphoma (EBV), cervical cancer (HPV)
, non-Hodkins lymphoma (EBV), cervical cancer (HPV)")
22
Secondary Immunodeficiency
Acquired Immunodeficiency Syndrome Treatment options Nucleoside reverse transcript inhibitors Non-nucleoside reverse transcript inhibitors Protease inhibitors Entry/fusion inhibitors (gp120, gp41, chemokine receptors) Integrase inhibitor
 Integrase inhibitor.")
23
Pneumocystis pneumonia

24
HSV and Kaposi’s Sarcoma

25
Toxoplasmosis and Cryptosporidium

26
Secondary Immunodeficiency
Either due to loss of immunoglobulins Nephrotic syndrome Protein-losing enteropathy Or due to impaired synthesis of immunoglobulins and/or cells Severe malnourishment Destruction of bone marrow (e.g. lymphoproliferative disorders) Viral infections (CMV, measles, mono, et al.) Iatrogenic immunosuppression
 Viral infections (CMV, measles, mono, et al.) Iatrogenic immunosuppression.")
27
Organ Transplantation
Normal immunity helps protect the body from invasion by microorganisms and provides surveilance for development of cancer These same defense mechanisms will attack “foreign” transplanted tissue

28
Organ Transplantation
Successful organ transplantation requires Sufficient pharmacologic suppression of the immune system to prevent rejection Without oversuppression which would predispose patients to opportunistic infections, tumors and graft-versus-host disease

29
Organ Transplantation
Human leukocyte antigens (HLA), a.k.a MHC proteins, are strongly antigenic One gene is inherited from each parent for each HLA class (MHC I- A, B, C and MHC II- DP, DQ, DR) So a cell may express up to 12 different HLA proteins A, B and DR are the most important
, a.k.a MHC proteins, are strongly antigenic. One gene is inherited from each parent for each HLA class (MHC I- A, B, C and MHC II- DP, DQ, DR) So a cell may express up to 12 different HLA proteins. A, B and DR are the most important.")
31
Before Transplantation
Find organ donor who matches as many HLA alleles as possible Depending on organ, may need to use ABO (blood type) compatible organ as well Screen recipient for presence of existing antibodies to foreign HLA types Find suitable live donor (kidney, liver, lung, bone marrow) or cadaveric donor (above plus heart, pancreas)
 compatible organ as well. Screen recipient for presence of existing antibodies to foreign HLA types. Find suitable live donor (kidney, liver, lung, bone marrow) or cadaveric donor (above plus heart, pancreas)")
32
After Transplantation
Immunosuppression with one or more drugs Cyclosporine & tacrolimus- blocks action of the phosphatase (caclineurin) which normally turns on IL-2 production Sirolimus- blocks action of a kinase necessary for T-cell proliferation (and activation) Azathioprine & mycophenolate inhibit DNA synthesis Monoclonal antibodies (end in “ab”, Muromonab) bind to T-cell proteins and lead to destruction by other immune cells or bind to and block IL-2 receptor Corticosteroids increase synthesis of proteins which inhibit transcription of multiple cytokines (IL-2, et al.)
 which normally turns on IL-2 production. Sirolimus- blocks action of a kinase necessary for T-cell proliferation (and activation) Azathioprine & mycophenolate inhibit DNA synthesis. Monoclonal antibodies (end in ab , Muromonab) bind to T-cell proteins and lead to destruction by other immune cells or bind to and block IL-2 receptor. Corticosteroids increase synthesis of proteins which inhibit transcription of multiple cytokines (IL-2, et al.)")
33
After Transplantation
Monitor for rejection Hyperacute antibodies to proteins on transplant are present at time of transplantation, rapid destruction (within minutes to hours) of transplantion Acute failure of immunosuppression so that antibodies develop in the weeks or months following transplantation Chronic immunosuppression can not (yet) be perfect, small amounts of damage accumulate over years and eventually destroy transplant
 of transplantion. Acute failure of immunosuppression so that antibodies develop in the weeks or months following transplantation. Chronic immunosuppression can not (yet) be perfect, small amounts of damage accumulate over years and eventually destroy transplant.")
34
After Transplantation
Rejection, direct Donor APCs do what they do, but interact with recipient CD4 T-cells which have entered the transplant Recipient CD4 cells recognize MHC II on donor APCs as foreign CD4 cells recruit (cytotoxic) CD8 T-cells and B-cells (which differentiate into plasma cells and make antibodies) CD8 cells mediate cytotoxicity via foreign MHC I (which is on all nucleated cells, importantly endothelial cells)
 CD8 T-cells and B-cells (which differentiate into plasma cells and make antibodies) CD8 cells mediate cytotoxicity via foreign MHC I (which is on all nucleated cells, importantly endothelial cells)")
35
After Transplantation
Rejection, indirect Donor HLA antigens either enter the blood stream or are carried by donor dendridic cells (APCs) to recipient lymphoid tissue Recipient CD4 T-cells recognize the foreign antigens, enter circulation, find their way to the transplant, and cause inflammatory response Again, damage to endothelium is as important as damage to organ itself
 to recipient lymphoid tissue. Recipient CD4 T-cells recognize the foreign antigens, enter circulation, find their way to the transplant, and cause inflammatory response. Again, damage to endothelium is as important as damage to organ itself.")
36
Graft-versus-host Disease
Highest risk after BMT, but can occur after any transplant, including transfusion Donor lymphocytes in the transplant or transfusion recognize the recipient as foreign, but the recipient fails to recognize the donor lymphocytes as foreign The recipient lymphocytes fail to act either because the donor cells do not look foreign to them or they are defective in number or function

37
A1 A7 A1 A7 A1 A7 A1 A7 A1 A3 A1 A7 A1 A7 A1 A7

38
A1 A7 A1 A7 A1 A7 A1 A7 A1 A7 A1 A7

39
A1 A7 A1 A7 A1 A7 A1 A7 ZZZZZ… A1 A1 A7 A1 A7 A1 A7

40
A1 A7 A1 A1 A7 A1 A7 ZZZZZ… A1 A1 A7 A1 A7 A1 A7

41
A1 A7 A1 A1 A1 A7 A1 A1 A7 A1 A7

42
Graft-versus-host Disease
Any tissue can be affected, but liver, skin and GI tract are particularly affected Present with jaundice, rash and/or bloody diarrhea May be acute with rapid increase of symptoms or chronic with insidious progression Treat with increased immunosuppression and/or immunomodulation (photopheresis)
")
46
Questions?

Similar presentations










 –Phagocytic cells: WBC’s –Natural killer cells: perforins –Resident bacteria.>")






. Immune Deficiency Disorders Immunodeficiencies can be divided into primary immunodeficiency disorders, and secondary immunodeficiency.>")





