Download presentation
Presentation is loading. Please wait.

1
DR.S.M.THIRUNUAVUKKARASU MD., DEPARTMENT OF MEDICINE
GOVT. THENI MEDICAL COLLGE THENI.

2
My Sincere Thanks To Our Beloved DEAN
THIS COLLEGE and MY TEACHERS AND DEAR JUNIORS., Who Encouraged Me To Present This Rare Case

3
My sincere thanks to Our
Beloved DEAN THIS COLLEGE and MY TEACHERS AND DEAR JUNIORS., Who Encouraged Me To Present This Rare Case

8
antenatal USG Screening of mothers and its sequeale if neglected
For this 6th chance to stand before YOU TO ENLIGHTEN THE Importance of antenatal USG Screening of mothers and its sequeale if neglected

9
The indication for LSCS – Previous LSCS ANTENATAL CARE – Adequate
This is an interesting case history Of a baby born by elective repeat LSCS at a Pvt. Hospital in our Dt The indication for LSCS – Previous LSCS ANTENATAL CARE – Adequate USG – 4 times at regular intervals (BOTH BY RADIOLOGIST AND NON RADIOLOGIST). LMP EDD G2P1L1-HIV &VDRL-NR
. LMP EDD G2P1L1-HIV &VDRL-NR.")
10
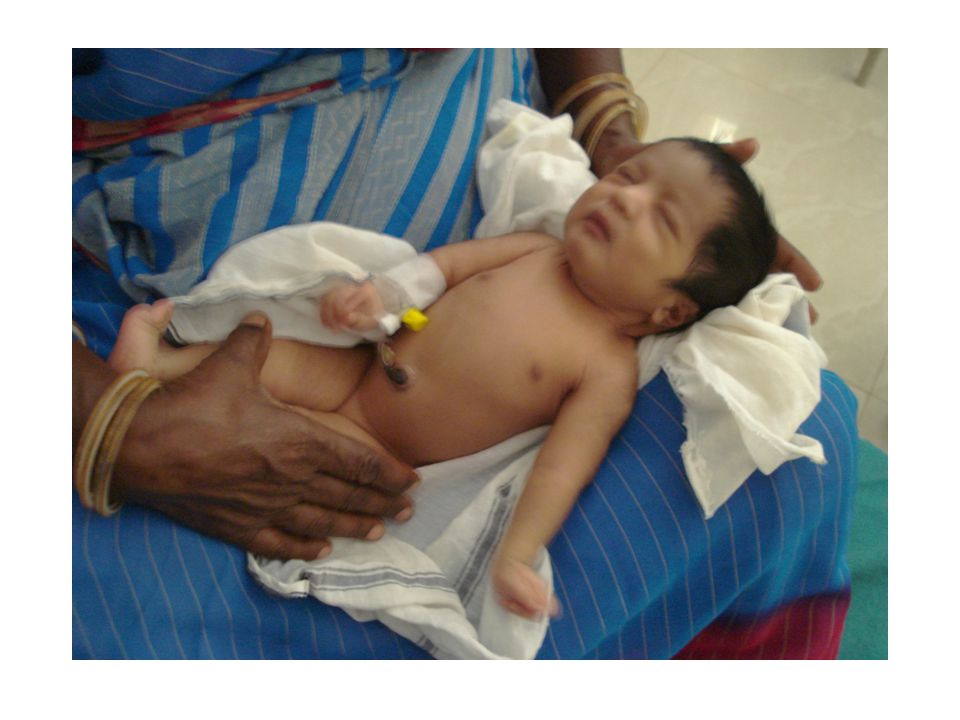
Do-LSCS(with s) PM Birth wt Kgms., Term female baby Not cried immediately after birth Resuscitation failed Passed urine and meconium Had poor respiratory effort and suspected to have RDS and Multiple cong. Anomalies.

11
This baby was referred to our Hospital 4 hours after birth by a Peadiatrician-reached our hospital within 6 hours of birth: Admitted in SNN ward – Thanks to DR.V.SEENIVASAN (efforts to arrive the probable diagnosis)
")
12
DORSIFLEXION OF BOTH FEET. SINGLE PALMAR CREASE
HIS OBSERVATION - DORSIFLEXION OF BOTH FEET. SINGLE PALMAR CREASE FLEXION CONTRACTURE OF FINGERS AND RESTRICTED FLEXION OF BOTH WRIST LT. CONGENITAL CATARACT ANAL OPENING PATULOUS,CRANIOSYNOSTOSIS DIAGNOSIS- LATE PRETERM- AGA- RD MULTIPLE CONGENITAL ANAMOLIES LT. CONGENITAL CATARACT- CRANIOSYNOSTOSIS ?ORTHROGRYPOSIS CONGENITA (10.30PM )
")
13
INVESTIGATIONS- Hb:10.8 grams., Dc –P47%E3%L50% Blood grouping- O -+ X Ray chest- ?Cardiomegaly?Thymic shadow

14
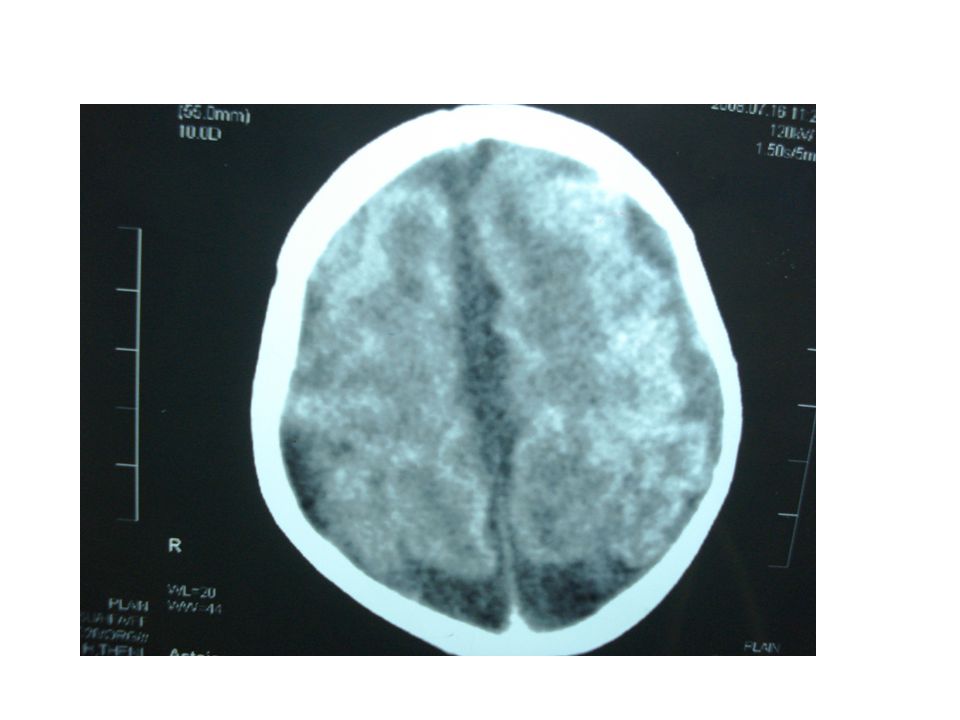
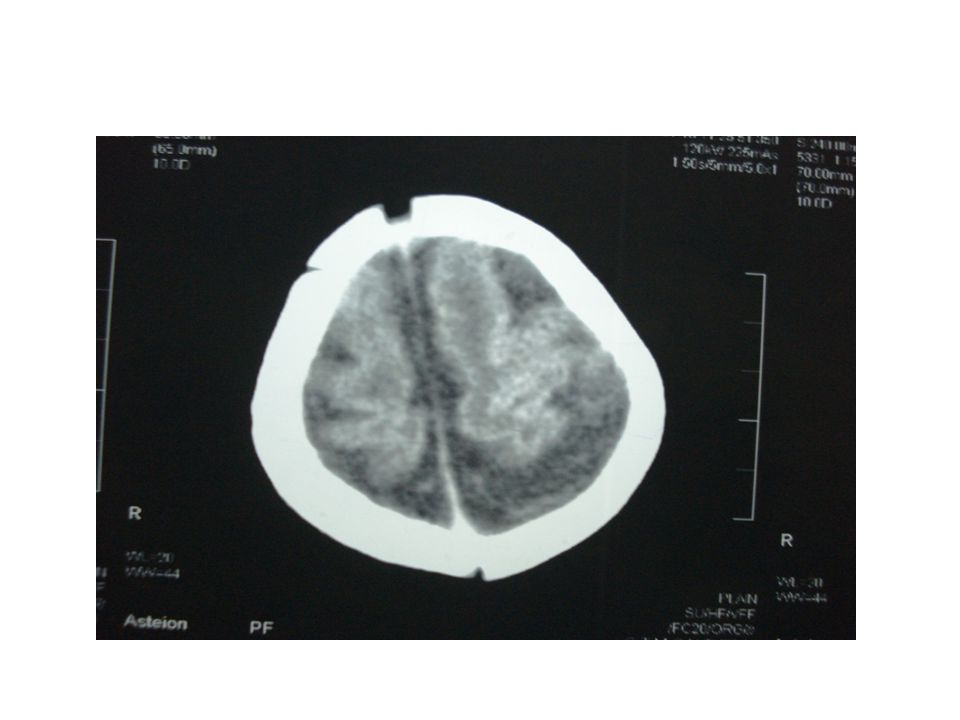
: Neurosonogram All ventricles are dilated Measures 19 mm at the atrium level Promenent cisterna magna Advised to take CT

15
CT brain – cerebral atrophy with prominent SA space with ventriculomegaly. Post fossa N MRI advised

31
:Echo was taken Ias and Ivs intact Lt to Rt shunt with small PDA

32
With Hypotonia + 19.7.08:ORTHO opinion 1 wk old baby
With B/L extended knees With B/L Calcaneo valgus deformity With Hypotonia + DIAGNOSED – ARTHOGRPOSIS MULTIPLEX CONGENITA

33
Discussion – Synonyms and related keywords. Arthrogryposis multiplex congenita AMC Multiple congenital contractures Multiple congenital joint contractures Fetal akinesia Decreased fetal movements

34
Arthrogryposis multiplex congenita
is a Heterogenous group of disorders Characterized by multiple joint contractures of prenatal onset with deformed, rigid joints, found throughout the body at birth.

35
Causes Extrinscic causes-oligohydramnios twinning uterine masses
Intrinsic causes - Neurogenic, myogenic, skeletal, connective tissue abnormalities,

36
Pathophysiology- The major cause of arthrogryposis is
fetal akinesia(decreased fetal Movements), Maternal disorders: (IU infection, drugs, trauma, maternal illness) Generalized fetal akinesia:- can also lead to polyhydramnios, pulmonary hypoplasia, micrognathia, occular hypertelorism, short umblical cord
, Maternal disorders: (IU infection, drugs, trauma, maternal illness) Generalized fetal akinesia:- can also lead to. polyhydramnios, pulmonary hypoplasia, micrognathia, occular hypertelorism, short umblical cord.")
37
During early embryo genesis
Joint development is almost always normal. motion is essential for the normal development of joints and their contiguous structures: lack of fetal movements causes extra connective tissue to develop around leading to joint fixation and movement limitation and aggravation of the joint contractures, with dislocation of joints (hip, knee)..
..")
38
Most cases are Neurogenic in origin,
as a result from congenital/acquired defect in the organization or number of anterior horn cells, roots, peripheral nerves or motor end plates

39
Producing muscular weakness and resultant joint immobility at critical
stages of intrauterine development. Pattern of deformity Type I –VIII. Myopathic Multiple congenital contractures account for 10% and AR transmision. Limited intrauterine movement is the common feature to all types of arthrogryposis. ..

40
NORMAL FETAL MOVEMENTS:
3 or more discrete body/limb movements In 30 min observation or less ( episodes of active continuous movement considered as single movement)
")
41
a fibrous ankylosis Histologically –
muscle mass will be small with fibrosis and fat between muscle fibers. The periarticular soft tissue structures are fibrotic and create a fibrous ankylosis

42
Incidence: – 1 in 3,000 live births. Race:-No racial predilection Sex:–males are primarily affected in x-linked recessive disorders, otherwise both are equally affected. Age:– is usually detected at birth or in utero by usg.

43
How to diagnose AMC ? X –RAY CT SCAN MRI EMG &NCS SERUM ENZYME TESTS MUSCLE BIOPSY WHAT ELSE WE NEED?

44
Clinical examination best modality for establishing the diagnosis
remains the best modality for establishing the diagnosis Rest of the above said things are helpful In assessing the invovlement of the Skeletal system (hip dislocation,scoliosis) Scoliosis has been reported in 10-30%
 Scoliosis has been reported in 10-30%")
45
History Review hyperextensibility, dislocation of joints, clubfeet in others. Consanguity: is more common in families with rare recessive diseases. Increased maternal and paternal age increases the possibility. Pregnancy history: 1. Infants born to mothers with myotonic dystrophy, myasthenia gravis, or multiple sclerosis are at risk of having a child with AMC. 2. Maternal infections can lead to CNS & PN destruction with secondary congenital contractures. 3. Maternal hyperthermia of more than 39* C for an extended period can cause contractures due to abnormal nerve growth or migration. 4. Exposure to teratogens. 5. Chronic amniotic fluid leakage may cause fetal constraint and contractures 6. Uterine abnormalities: bicornuate/septate uterus or fibroid. 7. Antenatal bleeding, threatened abortion, attempted or failed termination, abdominal trauma and abnormal fetal lie. -

46
Physical examination-
Joint contractures and clinical manifestations may vary from case to case Some of the common characters are Involved extremities are fusiform or cylindrical in shape with thin subcutaneous tissue and absent skin creases Deformities are usually symmetrical and increasing severity distally with the hands and feet typically the most deformed. Joint rigidity with Muscles atrophy or muscle groups may be absent

47
Sensation may be usually normal DTR diminished or absent.

48
Lab studies- In general lab studies are not extremely useful.

49
is the only helpful tool- at least to study
Imaging studies- Photography- to document the extent of deformities and to evaluate the skeletal and joint abnormalities. CT- scan- to evaluate the CNS and the muscle. MRI- to evaluate the muscle mass obscured by contractures USG- prenatal usg is the only helpful tool- at least to study the decreased fetal movements abnormal fetal lie & Poly/Oligohydramios

50
Managements- Medical care- no completely successful
approach to treatment has been found. 2. Early vigorous physical therapy to stretch contractures is very important in improving joint motion and avoiding muscle atrophy. 3. Feeding assistance and intubation for pts. with severe trismus. 4. Surgical care properly sequenced corrective surgical procedures to maximize function.

51
Prevention & Recurrence
Recurrence risk depends on whether the contractures are extrinsically or intrinsically derived. Extrinsically derived contractures have a low recurrence risk. Intrinsically derived contractures have risk of recurrence depends on etiology. AMC may he inheritied in the following ways. AD- RR IS 50%--- AR- RR IS 25% (both parents are obligatory carriers.) x-LR- all daughters of affected males are carriers (sons have 50% chance of affection- daughters have 50% chance of being carriers.) Sporadic/mitochondrial mutations/ multifactorial
 x-LR- all daughters of affected males are carriers. (sons have 50% chance of affection- daughters have 50% chance of being carriers.) Sporadic/mitochondrial mutations/ multifactorial.")
52
Complications – Per operative issues- difficulties with airway
management, problematic intravenous access, intra operative hyperthermia. 2. Problems in intubations- small jaw, limited tm joint movement, narrow airway. 3. Osseous hypoplasia due to decreased mechanical use in developing bone which are prone to fracture at multiple sites.

53
Prognosis- Neonates require ventilator –poor prognosis
(predictors- decreased fetal movements, Polyhydramnios, micrognathia,thin ribs,) delayed developmental milestones 2. Skeletal changes secondary to original deformities may worsen the patients overall condition. 3. Extrinsically derived contractures - carry good prognosis 4. Intrinsically derived contractures – not so
 delayed developmental milestones. 2. Skeletal changes secondary to original deformities. may worsen the patients overall condition. 3. Extrinsically derived contractures - carry good prognosis. 4. Intrinsically derived contractures – not so.")
54
Family(patient) education
The birth of a child with AMC may be a catastrophic event for parents and family. They may experience anger, feelings of guilt, repulsion , disappointment, depression. Family members may have difficulty in understanding or accepting the diagnosis. Family members may also be informed about additional unrecognized malformations, risk of MR, the risk of recurrence.- Why -

55
BEFORE TO DO STERILIZATION TRY TO RULE OUT(keep in mind) MAJOR ANAMOLIES ( chromosomal anamolies imperforate anus ambigous genitalia ext. urethral meatal anamolies) In born errors of metabolism(later)
 In born errors of metabolism(later)")
56
THANKYOU SMT ARASU

Similar presentations




























 Unlike: neuronopathies: secondary.>")


., DMRD., D.Diab., F.Echo., DEPARTMENT OF MEDICINE GOVT. THENI MEDICAL COLLGE,THENI.>")


