Download presentation
Presentation is loading. Please wait.

1
Lymphomas: Molecular basics, terms and definitions
Dr Epari Sridhar Asst Professor Pathology TMC

2
Lymphoid neoplasms Classification requires multiparameter approach
Clinical features Morphology Immunophenotyping and Molecular methods, in some Both diagnostic and prognostic significance

3
Lymphomas – molecular testing - Utility
Demonstration of a clonality reactive vs neoplastic proliferation Aid in correct lymphoma diagnosis Inconclusive histologic and immunophenotypic data Useful for classification, staging, and prognostication Information to guide appropriate choice of therapy Evidence of remission or relapse. Identify disease-associated findings such as an associated virus specific chromosomal translocation, that is useful in subclassification.

4
Lymphomas – Molecular testing - Targets
Antigen receptor gene rearrangements Non-random chromosomal abnormalities Translocations Numerical aberrations

5
Terms Karyotype refers to a full set of chromosomes from an individual
Chromosome anomaly, abnormality or aberration reflects an atypical number or a structural abnormality in one or more chromosomes. Two basic groups: Numerical and structural anomalies.

6
Chromosomal Numerical Anomaly
Aneuploidy: abnormal number of chromosomes Monosomy: chromosome missing from a pair. Denoted as ‘Ms’ Trisomy, tetrasomy etc: More than two chromosomes of a pair. ‘Ts’ for trisomy and ‘Tet’ for tetrasomy

7
Chromosomal structural abnormalities
Deletions: A portion of the chromosome is missing or deleted. Denoted as symbol ‘del’ Terminal Deletion - a deletion that occurs towards the end of a chromosome. Intercalary Deletion / Interstitial Deletion - a deletion that occurs from the interior of a chromosome. Microdeletions: An extremely small amount of a chromosome is missing, possibly only a single gene. Duplications (dp/dup): Portion of the chromosome is duplicated, resulting in extra genetic material. Gene duplications or amplification Translocations: A portion of one chromosome is transferred to another (nonhomologous) chromosome.
: Portion of the chromosome is duplicated, resulting in extra genetic material. Gene duplications or amplification. Translocations: A portion of one chromosome is transferred to another (nonhomologous) chromosome.")
8
Chromosomal translocations
Two main types of translocations: Reciprocal (non-Robertsonian) translocation: segments from two different chromosomes have been exchanged. Robertsonian translocation: an entire chromosome has attached to another at the Centromere. Balanced: even exchange of material with no genetic information extra or missing and ideally with full functionality Unbalanced: Unequal exchange of material resulting in extra or missing genes.
 translocation: segments from two different chromosomes have been exchanged. Robertsonian translocation: an entire chromosome has attached to another at the Centromere. Balanced: even exchange of material with no genetic information extra or missing and ideally with full functionality. Unbalanced: Unequal exchange of material resulting in extra or missing genes.")
9
Chromosomal translocations - Denotation
The International System for Human Cytogenetic Nomenclature (ISCN) t(A;B)(p1;q2) ‘t’ stands for translocation (A;B) denotes a translocation between chromosome A and chromosome B. (p1;q2) denotes precise location within the chromosome for chromosomes A and B respectively—with p indicating the short arm of the chromosome, q indicating the long arm, and the numbers after p or q refers to regions, bands and sub-bands Examples: Burkitt lymphoma: t(8;14)(q24;q32) Mantle cell lymphoma: t(11;14)(q13;q32) Follicular lymphoma: t(14;18)(q32;q21)
 t(A;B)(p1;q2) ‘t’ stands for translocation. (A;B) denotes a translocation between chromosome A and chromosome B. (p1;q2) denotes precise location within the chromosome for chromosomes A and B respectively—with p indicating the short arm of the chromosome, q indicating the long arm, and the numbers after p or q refers to regions, bands and sub-bands. Examples: Burkitt lymphoma: t(8;14)(q24;q32) Mantle cell lymphoma: t(11;14)(q13;q32) Follicular lymphoma: t(14;18)(q32;q21)")
10
Chromosomal structural abnormalities
Inversion: A portion of the chromosome has broken off, turned upside down and reattached, therefore the genetic material is inverted without loss of genetic information. Denoted as symbol ‘in’ Paracentric: Do not include the centromere and both breaks occur in one arm of the chromosome. Pericentric: include the centromere and there is a break point in each arm. Ring chromosome: A portion of a chromosome has broken off and formed a circle or ring. This can happen with or without loss of genetic material. denoted by the symbol ‘r’ Isochromosome: Formed by the mirror image copy of a chromosome segment including the centromere. denoted by the symbol ‘iso’

11
Chromosomal structural abnormalities
Insertion: On a chromosomal level, refers to the insertion of a larger sequence into a chromosome. On a genetic (gene) level is the addition of one or more nucleotide base pairs into a DNA sequence. Can be anywhere and of any size incorrectly inserted into a DNA sequence of one chromosome inserted into another. e.g.,Is(7;1) - insertion of part of Chr 7 into Chr 1
 level is the addition of one or more nucleotide base pairs into a DNA sequence. Can be anywhere and of any size incorrectly inserted into a DNA sequence of one chromosome inserted into another. e.g.,Is(7;1) - insertion of part of Chr 7 into Chr 1.")
12
Other Human Chromosome Nomenclature
Symbols used to designate these whole arm chromosome changes are: "+" to indicate the presence of a specific additional autosome "–" to indicate the absence of a specific autosome "O" to indicate a missing sex chromosome Additional Xs or Ys to indicate supernumerary sex chromosomes Number of chromosomes is specified, followed by a comma and a specification of the whole arm chromosome change.

13
Chromosomal structural abnormalities
Paracentric inversion Pericentric inversion Isochromosome Insertion Ring chromosome

14
Lymphomas – Molecular genetic methods
Karyotyping Limited use, especially in lymphomas Difficult to get adequate cell growth esp. LGNHL Cannot detect IgH and TCR re-arrangements Southern blot analysis Traditional gold standard for most molecular diagnostic testing. Requires fresh tissue in fairly large amounts Labor-intensive, time-consuming method. Requires large percentage of abnormal cells in the sample (5–10%) Polymerase chain reaction (PCR) methods Direct PCR and Reverse transcriptase (RT) – PCR In-situ hybridisation (ISH) Fluorescence in situ hybridization (FISH) Chromogenic in-situ hybridisation (CISH), Silver in-situ hybridisation (SISH) and Rapid in-situ hybridiation (RISH) In-situ PCR PCR in the cell on a slide, and visualized in the same way as in traditional ISH Technically difficult, is often inconsistent, Not used in most diagnostic laboratories. Others – CGH, Spectral karyotyping, Micro-array technology
 Polymerase chain reaction (PCR) methods. Direct PCR and Reverse transcriptase (RT) – PCR. In-situ hybridisation (ISH) Fluorescence in situ hybridization (FISH) Chromogenic in-situ hybridisation (CISH), Silver in-situ hybridisation (SISH) and Rapid in-situ hybridiation (RISH) In-situ PCR. PCR in the cell on a slide, and visualized in the same way as in traditional ISH. Technically difficult, is often inconsistent, Not used in most diagnostic laboratories. Others – CGH, Spectral karyotyping, Micro-array technology.")
15
Lymphomas – molecular testing –targets
Antigen receptor gene re-arrangements – Ig (Igk, Igλ and IgH) & TCR (TCRγ, TCRβ, TCRα/δ) Southern blot analysis Fresh tissue Slow turn –around time Labour intensive Low analytical sensitivity PCR methods Preferred first-line approach Almost replaced the SB analysis as requires less tissue and permissible with FFPE tissues Chromosomal translocations and aneusomies: DNA based and RNA transcripts (fusion genes) Preferred methods: PCR and FISH Conventional cytogenetics
 & TCR (TCRγ, TCRβ, TCRα/δ) Southern blot analysis. Fresh tissue. Slow turn –around time. Labour intensive. Low analytical sensitivity. PCR methods. Preferred first-line approach. Almost replaced the SB analysis as requires less tissue and permissible with FFPE tissues. Chromosomal translocations and aneusomies: DNA based and RNA transcripts (fusion genes) Preferred methods: PCR and FISH. Conventional cytogenetics.")
16
Antigen receptor re-arrangement
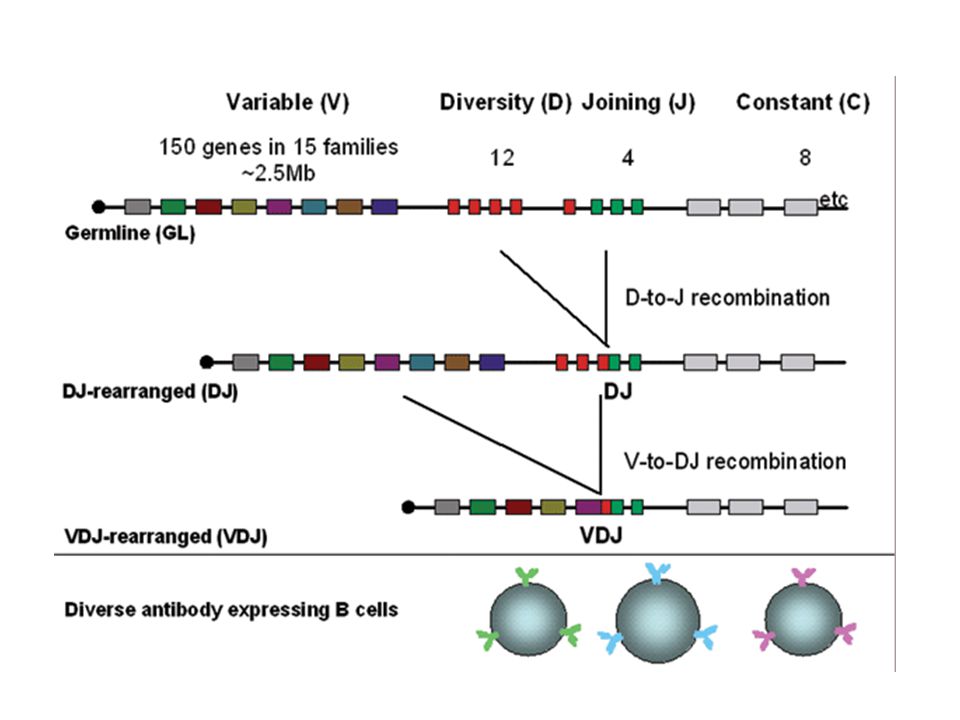
Ig and TCR genes – discontinuous segments that encode for the variable (V), joining (J), constant (C) and sometimes diversity (D) regions Diverse antigen detection capability is generated by different synergistically acting mechanisms: Somatic recombination Complementarity-determining regions (CDRs) In-Frame alignment of gene segments Genetic hierarchy Allelic exclusion Class switching
, joining (J), constant (C) and sometimes diversity (D) regions. Diverse antigen detection capability is generated by different synergistically acting mechanisms: Somatic recombination. Complementarity-determining regions (CDRs) In-Frame alignment of gene segments. Genetic hierarchy. Allelic exclusion. Class switching.")
18
Clonality assays – PCR vs Southern blot
DNA amount 1 µg or less 30 µg min. per probe DNA quality/size Can be severely degraded, bp DNA High quality, HMW DNA needed, atleast 20 kb DNA source Fresh or frozen or PBs Fresh or frozen Restricition enzyme digestion Not needed Required Gel electrophoresis Polyacrylamide gels, denaturing gradient gels & non-gel based methods Agarose gel required Time 1 to 2 days 1 to 2 weeks Detection methods Fluorescent dyes, silver stain, chemiluminescence, radioactivity Usually radioactivity, less often chemiluminescence. Senstivity 1 cell per 103 cells 1%-5% of total DNA False negative Common for B-cell lymphomas; uncommon in T-cell lymphomas Rare

19
Polymerase chain reaction (PCR)
In-vitro amplification of specific DNA sequences by primer extension of complementary strands of DNA Amplifies a single or few copies of a piece of DNA across several orders of magnitude, generating thousands to millions of copies of a particular DNA sequence Presently, the most preferred first-line approach in the molecular diagnostic tool

20
PCR Cycle Denaturation 95-98ºC Annealing 50-65ºC Elongation 75-80ºC 5’
3’ 5’ Denaturation 95-98ºC Annealing 50-65ºC Elongation 75-80ºC

21
PCR Cycle

22
Exponential Amplification
No. of Thermal Cycles Copies of Target (PCR Products = Amplicons) 1 2 4 3 8 16 5 32 6 64 20 1,048,576 30 1,073,741,824 (1x109)
 ,048, ,073,741,824 (1x109)")
23
PCR Methodology DNA –based and m-RNA based
AgR rearrangements by DNA based Gene fusion – m-RNA based Qualitative vs quantitative assays Most diagnostic assays are qualitative: simply detect presence or absence Quantitative – required for MRD Assays and detection systems Assay design Single primer set vs hemi-nested vs nested Monoplex vs multiplex reactions Primer design Consensus vs gene family specific

24
Multiplex-PCR Molecular equivalent of multitasking
Several pairs of primers annealing to different target sequences. Permits the simultaneous analysis of multiple targets in a single sample. Multiplex Ligation-dependent Probe Amplification (or MLPA): multiple targets to be amplified using only a single pair of primers.
: multiple targets to be amplified using only a single pair of primers.")
25
Nested PCR Increases the specificity of DNA amplification and more successful in specifically amplifying long DNA products. Two sets of primers are used in two successive reactions. In the first PCR, one pair of primers is used to generate DNA products, which may contain products amplified from non-target areas. The products from the first PCR are then used as template in a second PCR, using one ('hemi-nesting') or two different primers whose binding sites are located (nested) within the first set, thus increasing specificity.
 or two different primers whose binding sites are located (nested) within the first set, thus increasing specificity.")
26
Quantitative PCR (Q-PCR)
Measures the specific amount of target DNA (or RNA). Special thermal cyclers are used that monitor the amount of product during the amplification. Quantitative Real-Time PCR (QRT-PCR): measures the amount of amplified product by using fluorescence dye tagged primers.
. Special thermal cyclers are used that monitor the amount of product during the amplification. Quantitative Real-Time PCR (QRT-PCR): measures the amount of amplified product by using fluorescence dye tagged primers.")
27
Reverse Transcription PCR ( RT-PCR)
Reverse transcribe and amplify RNA to DNA. Before the PCR reaction, conversion of RNA to cDNA is done by a reverse transcriptase enzyme.

28
Methylation-specific PCR (MSP)
Identifies patterns of DNA methylation at CpG (cytosine-guanine) islands. Bisulphite conversion - converts unmethylated cytosine bases to uracil, which is complementary to adenosine in PCR primers. Two amplifications are then carried out: One primer set anneals to DNA with cytosines (corresponding to methylated cytosine), and Other set anneals to DNA with uracil (corresponding to unmethylated cytosine).
 islands. Bisulphite conversion - converts unmethylated cytosine bases to uracil, which is complementary to adenosine in PCR primers. Two amplifications are then carried out: One primer set anneals to DNA with cytosines (corresponding to methylated cytosine), and. Other set anneals to DNA with uracil (corresponding to unmethylated cytosine).")
29
PCR Methodology - Product detection system
Simple gel electrophoresis Most frequently employed Based on size - agarose and polyacralymide Requires ethidium bromide staining and UV illumination Hybridisation with labelled probes PAGE allows superior resolution and preferred for small PCR products Cannot achieve single base resolution Not quantitative Insensitive in detecting small monoclonal popln esp. in the background of polyclonal population

30
PCR Methodology – Other detection systems
Complex gel electrophoresis Denaturing gradient gel electrophoresis (DGGE) Temperature gradient gel electrophoresis (TGGE) Heteroduplex analysis in mutation detection enhancing gels Single strand conformational polymorphism analysis (SSCP) Solution based methods Colorimetric, fluorescent and chemiluminscent Less commonly used Capillary electrophoresis with automated fluorescent DNA fragment analysis (CEGS; GeneScan) Considered to superior of all because of high sensitivity and high throughput Method of choice
 Temperature gradient gel electrophoresis (TGGE) Heteroduplex analysis in mutation detection enhancing gels. Single strand conformational polymorphism analysis (SSCP) Solution based methods. Colorimetric, fluorescent and chemiluminscent. Less commonly used. Capillary electrophoresis with automated fluorescent DNA fragment analysis (CEGS; GeneScan) Considered to superior of all because of high sensitivity and high throughput. Method of choice.")
31
Heteroduplex analysis
Sequence variations in dsDNA can cause bends in the double helix, or even alter the basic structure of the helix and thus restricts the mobility of the same in the media. A mismatch between the two strands of DNA in a duplex can produce a more radical kink in the structure, producing a heteroduplex species which can easily be resolved from the homoduplex by electrophoresis. heteroduplex products will result in a smear of slow migrating products

32
PCR - techniques Indications Suitable specimens:
AgR gene rearrangements Specific translocations MRD detection and monitoring Suitable specimens: Small tissue biopsies including BMBx & BMA, cells scraped/microdissected from slides and specimens Both fresh and FFPE tissue samples

33
PCR – techniques – laboratory factors
Significant interlaboratory variation in the senstivity of clonal detection with the paraffin fixed tissues Factors affecting Fixatives- formalin is the best; mercuric fixatives and Bouin fluid are least suitable Decalcification – EDTA is better than formic acid Methods of extraction of DNA

34
CEGS; Genescan Advantages Limitations
Obviates labour intensive gel preparation and exposure to hazardous UV light and ethidium bromide Speed : read out requires hardly 30 minutes Automatic data processing and electronic storage Achieves single base pair resolution Comparable sensitivity Limitations Highly expensive Not resistant for false positives

35
PCR – Sensitivity & Specificity
Case selection Nature of sample (nature of background cellularity) Type of detection methods used Specificity Lineage infidelity – well recognized in lymphoblastic lymphomas Clonality does not always equate with malignancy nor vice versa
 Type of detection methods used. Specificity. Lineage infidelity – well recognized in lymphoblastic lymphomas. Clonality does not always equate with malignancy nor vice versa.")
36
PCR – T Cell clonality testing - Indications
T-cell lymphomas are often difficult to diagnose Difficult to distinguish from the benign reactive T cell proliferations by immunophenotyping Molecular assay for clonality : targeting TCRγ and TCRβ receptor genes TCRγ is preferred due to the simplicity of the structure

37
PCR – T Cell clonality testing
TCRγ rearrangements Primers combination of Vγ and Jγ – detects all possible rearrangements - Four V regions and five J regions PAGE is good enough for separation of products and detection but Heteroduplex analysis and CEGS – best Qualitative sensitivity: Wide reported range (60-100%) With multiple primers PAGE achieves 80-90% and high resolution like heteroduplex analysis or CEGS can reach upto 100% Analytical sensitivity Routine PAGE – 1-5%; Much higher with CEGS Test specificity and positive predictive value Range from % Low in lymphobastic lymphomas, High false positivity in inflammatory dermatoses, sometimes in plasma cell dyscrasias and Hodgkin lymphomas
 With multiple primers PAGE achieves 80-90% and high resolution like heteroduplex analysis or CEGS can reach upto 100% Analytical sensitivity. Routine PAGE – 1-5%; Much higher with CEGS. Test specificity and positive predictive value. Range from % Low in lymphobastic lymphomas, High false positivity in inflammatory dermatoses, sometimes in plasma cell dyscrasias and Hodgkin lymphomas.")
38
CD24251: 19/F, c/o HL; CE17159:24/F, c/o HL
PCR for TCRγ gene rearrangement

40
PCR – T Cell clonality testing
TCRβ gene rearrangements Less often used due to complexity of the gene structure Difficult for consensus primers Qualitative sensitivity : range 50-80% Quantative sensitivity: 2-5% Clonal detection range may be increased by as much as 20% if used along with TCRγ

41
PCR – B Cell clonality testing
IgH gene testing is the principal approach Qualitative sensitivity: >50% to virtually 100% - depends upon case mix, primer details and detection methods False negatives are known to occur in FL, MZL and DLBCLs False positives reported in AITLs and PTCL Clinical utility for tests is extremely rare Especially in diagnosing composite B cell lymphomas for determining two different clones of B-cells

42
PCR assays - pitfalls Combination of technical and biological factors and interpretation errors False positive rates Contaminations Excessive amplification cycles Inorganic DNA extraction methods Pseuodoclonality: selective oligoclone amplification due to insufficient sample Inappropriate AgG rearrangements False negative rates Sampling errors DNA and RNA degradation PCR design Biological factors

43
By using complex multiplex assays with advanced methods of detection increases the chance of detection of clonality in benign/reactive conditions Molecular data should never be reported in isolation from all other clinicopathological factors in each case

44
Standardization of PCR assays
Multicentre European collaborative studies have been instituted to optimise and standardize the PCR assays for purpose of clonality studies in lymphoma clonality testing – Biomed 2 concerted action Involves the use of 107 standardised primers in a series of 18 multiplex reations Product detection either by heteroduplex or automated Genescan analysis.

45
Fluorescence in situ hybridization (FISH)
Allows detection of both structural and numerical chromosomal abnormalities Considered superior to PCR methods for detection of translocations and aneusomys Not widely used in the routine diagnostic evaluation of paraffin-embedded biopsies, Technically more demanding (perception) Uncertainties regarding diagnostic thresholds and result interpretation.
 Uncertainties regarding diagnostic thresholds and result interpretation.")
46
FISH Types Principle Probes: two types for translocation
Metaphase FISH Interphase FISH – for solid Txs and FFPET can be used Principle Visualization of bound of flourochlorome tagged DNA fragments to complementary target genomic region Probes: two types for translocation Dual fusion probes Superior due to lack of false positivity Break-apart Gives abnormal results for variant translocations also Do not detect the other gene involved Probes: two types for detection of copy number changes Locus specific Chromosome enumeration (centromeric or pericentromeric satellites)
")
47
Metaphase FISH Interphase FISH Courtesy: JMD November 2000, Vol. 2, No. 4

48
Normal cells Abnormal cells Schematic figs Dual fusion probes Break apart probes

49
A,B – Two different cases of Burkitt lymphoma showing fusion
signals for IgH/CMYC (as shown by arrows) A B
 A. B.")
50
FISH – Interpretation Acquire experience of normal and abnormal signal patterns for each probe applied, using negative tissues (eg. reactive lymph nodes) and relevant positive samples (eg. lymphomas known to contain the abnormality under investigation). Other factors to be aware of: the architecture of the tissue, including local variations in neoplastic cell content, fixation, and cellularity within the section; Nuclear truncation and the complex nature of genetic arrangements seen in some lymphoid neoplasms. Should have a HE stained slide at your hand.
 and relevant positive samples (eg. lymphomas known to contain the abnormality under investigation). Other factors to be aware of: the architecture of the tissue, including local variations in neoplastic cell content, fixation, and cellularity within the section; Nuclear truncation and. the complex nature of genetic arrangements seen in some lymphoid neoplasms. Should have a HE stained slide at your hand.")
51
FISH -Interpretation Choosing proper area for Evaluation
Preferably areas richest in abnormal cells bright, distinct signals and low background in which individual nuclei are clearly distinguishable But screening of entire area is essential For the presence of subclonal changes that might be of diagnostic and prognostic importance, e.g., the presence of t(8;14) only in a subpopulation might indicate transformation into a more aggressive lymphoma. Areas of nuclear overlapping with indistinct nuclear outlines and high cell density - should be avoided.
 only in a subpopulation might indicate transformation into a more aggressive lymphoma. Areas of nuclear overlapping with indistinct nuclear outlines and high cell density - should be avoided.")
52
Area to be avoided for interpretation

53
FISH -Interpretation Awareness of nuclear truncation artefacts induced by sectioning Should distinguish from loss of chromosome Establishment of cut off values for different probes and all signal patterns Complex chromsomal abnormalities

54
FISH - Applications Detection of numerical and structural chromosomal abnormalities Identification of marker chromosomes (rearranged chromosomes of uncertain origin) Detection of gene deletions and gene amplifications Detection of early relapse or minimal residual disease Identification of the origin of bone marrow cells following stem cell transplantation
 Detection of gene deletions and gene amplifications. Detection of early relapse or minimal residual disease. Identification of the origin of bone marrow cells following stem cell transplantation.")
55
FISH - Advantages Rapid technique, and large numbers of cells can be scored in a short period Efficiency of hybridisation and detection for is high for structural and numerical abnormalities Can be applicable in scant cellular specimens (post Tx samples and hypocellular samples Permits direct correlation of cytogenetic and morphologic features, enabling pathologists to differentiate malignant from benign conditions in equivocal cases

56
FISH - Limitations Restricted to those abnormalities that can be detected with currently available probes Only one or few abnormalities can be assessed simultaneously Cytogenetic data can be obtained only for the target chromosomes; Not a good screening tool for heterogenous diseases Requires fluorescence microscopy

57
Lymphomas - Gene expression profiling
Offers the prospects of future refining the lymphoma sub-classification at molecular level May provide prognostic data and potential for novel targeted therapies Presently a research tool and requires fresh tissue Technique: Co-hybridisation of differentially flourochrome labelled RNA or cDNA of tumour and normal tissue with a cDNA chip (lymphochip) The chips contain robotically arranged known cDNAs from hundreds to thousands of genes Confocal microscopy along with computerised image analysis system measure the emission spectra Signal intensity at each spot is proportional to the level of gene expression Large data generated can be investigated by using mathematical algorithims
 The chips contain robotically arranged known cDNAs from hundreds to thousands of genes. Confocal microscopy along with computerised image analysis system measure the emission spectra. Signal intensity at each spot is proportional to the level of gene expression. Large data generated can be investigated by using mathematical algorithims.")
58
Gene expression profiling - Utility
DLBCL 3 distinct subgroups based on differential expression of 1000 genes Germinal centre-like, activated B-cell like and 3rd distinct group, represents heterogenous group Germinal centre signature was shown to have better survival rates Further supervised analysis – five differential gene expression profiles Differential gene expression – early and late or advanced stages CLL Overexpression of ZAP 70 – aggressive course Mantle cell lymphoma Enables prediction of poor prognosis group Follicular lymphoma Tranformation to DCBCL characterized by altered gene expression profile Reports of prediction for response to rituximab therapy

59
Thank You

Similar presentations








 The order of the base pairs in the sequence of every human varies In a single.>")









 BIOTECHNOLOGY Lecture 7 5th May, 2006 PhD Course.>")

 FISH 2) “Restriction mapping” 3) Southern analysis : DNA 4) Northern analysis: RNA tells size tells which tissues or conditions.>")