Download presentation
Presentation is loading. Please wait.

1
Pediatric Soft Tissue Sarcomas
Michael Weintraub, M.D. Hadassah University Hospital Jerusalem, Israel

2
Cancer Types in Children
Leukemia CNS tumors Lymphoma – Hodgkin’s & non-Hodgkin’s lymphoma Neuroblastoma Wilms’ tumor Sarcoma – Bone (Ewing, osteosarcoma) Soft-tissue –Rhabdomyosarcoma, NRSTS Retinoblastoma Hepatic tumors Germ cell tumors
 Soft-tissue –Rhabdomyosarcoma, NRSTS. Retinoblastoma. Hepatic tumors. Germ cell tumors.")
3
Major Cancer Types in Children
Leukemia (CNS tumors) Lymphoma – Hodgkin’s & non-Hodgkin’s lymphoma (Neuroblastoma) Wilms’ tumor Sarcoma – Bone (Ewing, osteosarcoma) Soft-tissue –Rhabdomyosarcoma, NRSTS Retinoblastoma (Hepatic tumors) (Germ cell tumors)
 Lymphoma – Hodgkin’s & non-Hodgkin’s lymphoma. (Neuroblastoma) Wilms’ tumor. Sarcoma – Bone (Ewing, osteosarcoma) Soft-tissue –Rhabdomyosarcoma, NRSTS. Retinoblastoma. (Hepatic tumors) (Germ cell tumors)")
4
Nomenclature of Tumors
Tumors are named after their cell of origin and the embryonal layer that cell arose from The middle embryonal layer – the mesoderm- gives rise to mesenchymal tissues- bone, muscle, cartilage, adipose tissue, blood vessels and more Mesenchymal tumors are called sarcomas

5
Mesenchymal tumors Tumors of bone (Osteosarcoma, Ewing sarcoma)
Tumors of soft tissues (Soft tissue sarcomas=STS) Tumors of skeletal muscle (Rhabdomyosarcoma) Tumors of smooth muscle (Leiomyosarcoma) Tumors of adipose tissue (Liposarcoma) Tumors of fibroblasts (Fibrosarcoma) Tumors of cartilage (Chondrosarcoma, synovial sarcoma) Tumors of blood vessels (Angiosarcoma) MPNST, clear cell sarcoma, inflammatory myofibroblastic tumor, desmoid (fibromatosis), DSRCT, MFH
 Tumors of skeletal muscle (Rhabdomyosarcoma) Tumors of smooth muscle (Leiomyosarcoma) Tumors of adipose tissue (Liposarcoma) Tumors of fibroblasts (Fibrosarcoma) Tumors of cartilage (Chondrosarcoma, synovial sarcoma) Tumors of blood vessels (Angiosarcoma) MPNST, clear cell sarcoma, inflammatory myofibroblastic tumor, desmoid (fibromatosis), DSRCT, MFH.")
6
Pediatric soft tissue sarcomas
The most common form of soft-tissue sarcoma in childhood is rhabdomyosarcoma (50% of all STS) For convenience – all other soft-tissue sarcomas of childhood are called non-rhabdo soft tissue sarcomas (NRSTS) – and account for the remaining 50% of STS
 For convenience – all other soft-tissue sarcomas of childhood are called non-rhabdo soft tissue sarcomas (NRSTS) – and account for the remaining 50% of STS.")
7
Rhabdomyosarcoma

8
Rhabdomyosarcoma A tumor which arises from immature mesenchymal cells committed to skeletal muscle lineage RMS can arise in multiple organs giving rise to a wide spectrum of clinical presentations, therapeutic approaches and prognoses Some of these organs (e.g. – bladder) do not normally contain skeletal muscle
 do not normally contain skeletal muscle.")
9
Rhabdomyosarcoma - Epidemiology
Most common type of soft tissue sarcoma in children 3.5% of childhood cancer Incidence: 4.3/1,000,000 per year USA ~ 350 new cases/year; Ethiopia? ~ 150? Less? (Lower incidence of RMS in African-American girls and in Southeast Asia) 2/3 of cases occur in children < 6 years of age Genetic associations
 2/3 of cases occur in children < 6 years of age. Genetic associations.")
10
Cancer Types by Age Group
Tumor Type Ages 0-14 Ages 15-19 Leukemia 28% 10% CNS 22% Neuroblastoma 8% 0.2% NHL 6% Hodgkin’s 3.6% 16.8% Wilm’s tumor 0.3% Rhabdomyosarcoma 1.7% NRSTS 3.5% 5.1% Osteosarcoma 2.6% 4.2% Ewing sarcoma 1.5% 2.4% Germ cell/gonadal 12.4% Retinoblastoma 3.2% 0% Hepatoblastoma 1.3% Thyroid 1.1% 7.3% Melanoma 7.6%

11
Rhabdomyosarcoma - Epidemiology
Most common type of soft tissue sarcoma in children 3.5% of childhood cancer Incidence: 4.3/1,000,000 per year USA ~ 350 new cases/year; Ethiopia? ~ 150? Less? (Lower incidence of RMS in African-American girls and in Southeast Asia) 2/3 of cases occur in children < 6 years of age Genetic associations
 2/3 of cases occur in children < 6 years of age. Genetic associations.")
12
Genetics of Childhood cancer

13
Cancer – Pathogenesis I
Cancer is caused by the occurrence in a single, initial cell - of multiple genetic changes - “hits”- aberrations The genetic aberrations that lead to the transformation of a normal cell into a cancer (malignant) cell involve genes which regulate cell proliferation, differentiation and apoptosis (Proto-oncogenes, tumor suppressor genes) When a sufficient number of genetic “hits” have occurred in a single cell - that cell will have acquired the capacity to proliferate and metastasize – the “cancer cell”
 cell involve genes which regulate cell proliferation, differentiation and apoptosis (Proto-oncogenes, tumor suppressor genes) When a sufficient number of genetic hits have occurred in a single cell - that cell will have acquired the capacity to proliferate and metastasize – the cancer cell")
15
Cancer – Pathogenesis - II
In most human cancers, the changes in genes that control cell proliferation are not inherited but acquired (somatic changes) It is estimated that in order for a cell to transform into a cancer cell, changes must occur in 7-10 different genes For a single cell to accumulate a sufficient number of mutations takes time, and thus cancer is largely a disease of old age
 It is estimated that in order for a cell to transform into a cancer cell, changes must occur in 7-10 different genes. For a single cell to accumulate a sufficient number of mutations takes time, and thus cancer is largely a disease of old age.")
16
Cancer – Pathogenesis - III
If an individual inherits a mutation in one of the genes that control cell proliferation, than all the cells in that individual’s body have taken the first step in the path of malignant transformation The cells in the bodies of these individuals have a “head start” on the malignant process: They have a higher risk of developing tumors, and develop tumors at an earlier age The group of diseases in which individuals carry inherited/germline mutations in cancer genes are called cancer predisposition syndromes

19
Cancer predisposition syndromes (CPS)
In individuals with CPS only a very small fraction of the total cells in their body (or at risk organs) become neoplastic because other (somatic) mutations are required to develop a clinically detectable lesion (cancer phenotype) Individuals with CPS often develop multiple tumors that occur at an earlier age than in individuals whose cancer gene mutations have all occurred somatically (The head start) The tumor types are site specific (not all cancers are increased) – depending on the nature of the genetic “hit” Not all individuals with CPS will develop tumors – in fact – in many CPS – most will not (Down syndrome vs. RB)
 become neoplastic because other (somatic) mutations are required to develop a clinically detectable lesion (cancer phenotype) Individuals with CPS often develop multiple tumors that occur at an earlier age than in individuals whose cancer gene mutations have all occurred somatically (The head start) The tumor types are site specific (not all cancers are increased) – depending on the nature of the genetic hit Not all individuals with CPS will develop tumors – in fact – in many CPS – most will not (Down syndrome vs. RB)")
20
The role of heredity in childhood cancer
Most cancer cases in children do not have a hereditary basis - Leukemia – 2% Brain tumors – 1-3% Wilm’s tumor – 3-5% Retinoblastoma – 40% Optic gliomas – 45% Adrenocortical Carcinoma – 50-80% However – the exceptions are instructive

21
RMS in Cancer Predisposition syndromes
Cancer Types Beckwith-Wiedemann Wilms (60%, 20% bilateral), RMS, HB, NB, ACC, Gonadoblastoma; (7.5 % of patients develop cancer by age 8) Li-Fraumeni RMS, OS, glioma, Breast, Adrenal, leukemia 50% cancer incidence by age 30 (cf. 1% in general population) Costello syndrome RMS
, RMS, HB, NB, ACC, Gonadoblastoma; (7.5 % of patients develop cancer by age 8) Li-Fraumeni. RMS, OS, glioma, Breast, Adrenal, leukemia. 50% cancer incidence by age 30 (cf. 1% in general population) Costello syndrome. RMS.")
22
Rhabdomyosarcoma- Clinical Presentations

23
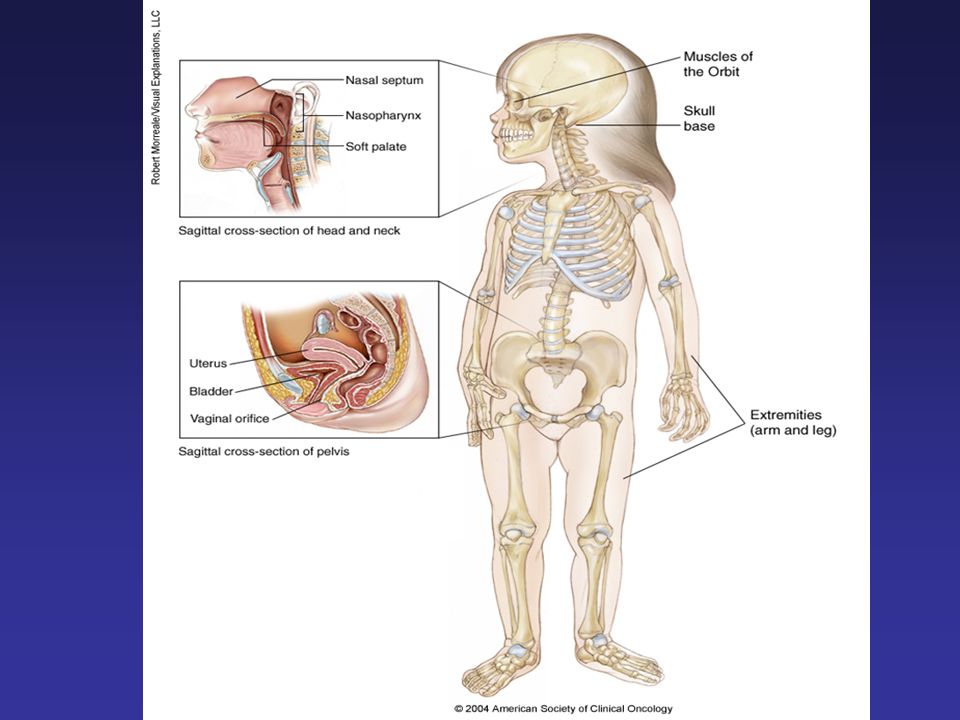
Rhabdomyosarcoma Sites of disease Head & Neck Orbit Parameningeal Non-Parameningeal Genitourinary Bladder Prostate Para-testicular Vagina/uterus Extremity Others

24
RMS – Clinical Presentation is Site Dependent
Orbit - Proptosis, ophthalmoplegia Other head and neck/parameningeal – nasal or aural obstruction, cranial nerve palsies Genitourinary tract – Bladder: Hematuria, urinary obstruction Paratesticular – painless scrotal mass Vaginal – Vaginal mass, discharge Extremities – Swelling, pain, lymph node involvement

25
Orbital rhabdomyosarcoma

26
Extremity RMS

28
Rhabdomyosarcoma – Approach to Diagnosis and Staging
Evaluation of primary site – XR, CT, MRI Biopsy / surgery Metastatic workup – CT chest, bone scan, bone marrow, PET

29
Rhabdomyosarcoma - Pathology
Two major histologic subtypes: I. Embryonal RMS (Botryoid and spindle cell variants) II. Alveolar RMS Undifferentiated sarcoma
 II. Alveolar RMS. Undifferentiated sarcoma.")
30
Poorly Differentiated Embryonal RMS difficult to distinguish from other small round blue cell tumors

31
Botryoid RMS

32
Alveolar RMS Small round cells floating in a pseudo-alveolar space representing fibrovascular septae

33
Small round blue cell tumors
Lymphoma Neuroblastoma Rhabdomyosarcoma Ewing/PNET Desmoplastic small round cell tumor (DSCRT) Poorly differentiated synovial sarcoma Small cell osteosarcoma
 Poorly differentiated synovial sarcoma. Small cell osteosarcoma.")
34
Small round blue cell tumors
Immunohistochemistry Electron microscopy Cytogenetics/Molecular Biology

35
Small round blue cell tumors Immunohistochemistry
Immunohistochemical markers Ewing / ESFT PAS+ (Glycogen); NSE (Neuron specific enolase); CD99; Fli1 Rhabdomyosarcoma Desmin, myosin, MyoD Lymphoma LCA=Leukocyte common antigen=CD45 specific markers CD30-HD,ALCL; CD20-B cell; CD3-T cell; TdT Neuroblastoma NSE; S100
; NSE (Neuron specific enolase); CD99; Fli1. Rhabdomyosarcoma. Desmin, myosin, MyoD. Lymphoma. LCA=Leukocyte common antigen=CD45. specific markers. CD30-HD,ALCL; CD20-B cell; CD3-T cell; TdT. Neuroblastoma. NSE; S100.")
36
Small round blue cell tumors
Immunohistochemistry Electron microscopy – features of muscle differentiation -= actin-myosin bundles, z-bands Cytogenetics/Molecular biology

37
Cytogenetics in Pediatric Solid tumors
Affected genes Embryonal rhabdomyosarcoma LOH 11p15 IGF-II Alveolar Rhabdomyosarcoma t(2;13)(q35;q14) t(1;13)(p36;q14) PAX3-FKHR PAX7-FKHR Neuroblastoma 1p36;17q; HSR;DM ?;?; N-myc Ewing sarcoma-PNET t(11;22)(q24;q12) t(21;22)(q22;q12) EWS-FLI1 EWS -ERG Malignant melanoma of soft parts t(12;22)(q13;q12) EWS-ATF1 Desmoplastic small round-cell tumor t(11;22)(p13;q11-12) EWS-WT1 Synovial sarcoma t(X;18)(p11.2;q11.2) SYT-SSX-1+2 Congenital fibrosarcoma and mesoblastic nephroma t(12;15)(p13;q25) ETV6-NTRK3
(q35;q14) t(1;13)(p36;q14) PAX3-FKHR. PAX7-FKHR. Neuroblastoma. 1p36;17q; HSR;DM. ; ; N-myc. Ewing sarcoma-PNET. t(11;22)(q24;q12) t(21;22)(q22;q12) EWS-FLI1. EWS -ERG. Malignant melanoma of soft parts. t(12;22)(q13;q12) EWS-ATF1. Desmoplastic small round-cell tumor. t(11;22)(p13;q11-12) EWS-WT1. Synovial sarcoma. t(X;18)(p11.2;q11.2) SYT-SSX-1+2. Congenital fibrosarcoma and mesoblastic nephroma. t(12;15)(p13;q25) ETV6-NTRK3.")
38
Rhabdomyosarcoma – Approach to Diagnosis and Staging
Evaluation of primary site – XR, CT, MRI Biopsy / surgery Metastatic workup – CXR/CT chest, bone scan, bone marrow, PET

39
Staging A process that defines the local and distant (metastatic) extent of a tumor Tumors have unique and consistent patterns of spread Wilms’ tumor to lungs and liver (not to bone or bone marrow) Neuroblastoma – bones, bone marrow, lymph, (not to lungs) Stage is associated with prognosis (metastatic disease is rarely curable)
 Neuroblastoma – bones, bone marrow, lymph, (not to lungs) Stage is associated with prognosis (metastatic disease is rarely curable)")
40
Wilms’ tumor - Staging Stage I II III IV V
Tumor confined to the kidney and completely resected. No penetration of renal capsule or sinus vessels. I Tumor extends beyond kidney but completely resected; a) penetration of renal capsule b) invasion of renal sinus c) biopsy d) local spillage during removal II Gross or microscopic residual (including gross spillage, positive margins, regional lymph nodes –renal hilar, para-aortic, or beyond, peritoneal implants, spillage beyond flank) III Metastatic disease outside abdomen (lungs, liver) IV Bilateral Wilms’ tumors V
 penetration of renal capsule b) invasion of renal sinus c) biopsy d) local spillage during removal. II. Gross or microscopic residual (including gross spillage, positive margins, regional lymph nodes –renal hilar, para-aortic, or beyond, peritoneal implants, spillage beyond flank) III. Metastatic disease outside abdomen (lungs, liver) IV. Bilateral Wilms’ tumors. V.")
41
Rhabdomyosarcoma – Evaluation of disease extent
Extent of disease in primary site – CT, MRI, PET Metastatic disease – Lungs, bones, lymph nodes Stage Clinical group (site and extent of resection)
")
42
Any site not considered favorable.
Term Definition Favorable site Orbit; non-parameningeal head and neck; genitourinary excluding kidney, bladder, and prostate; biliary tract. Unfavorable site Any site not considered favorable. T1 Confined to anatomic site of origin. T2 Extension and/or fixative to surrounding tissue. a Tumor ≤5 cm in maximum diameter. b Tumor >5 cm in maximum diameter. N0 No clinical regional lymph node involvement. N1 Clinical regional lymph node involvement. M0 No metastatic disease. M1 Metastatic disease.

43
Distant Metastasis Regional Lymph Nodes Tumor Size Sites of Primary Tumor Stage M0 N0, or N1, or NX Any size Favorable sites 1 N0 or NX T1a or T2a Unfavorable sites 2 N1 N0 or N1 or NX T1a, T2a, or T1b, T2b 3 M1 N0 or N1 Any site 4

44
I [Note: Approximately 13%]
Definition Group A localized tumor completely removed with pathologically clear margins and no regional lymph node involvement. I [Note: Approximately 13%] A localized tumor that is grossly removed with: (A) microscopic disease at the margin, (B) involved, grossly removed regional lymph nodes, or (C) both A and B. II [Note: Approximately 20% ] A localized tumor with gross residual disease after incomplete removal or biopsy only. III [Note: Approximately 48% ] Distant metastases are present at diagnosis. IV [Note: Approximately 18% ]
![I [Note: Approximately 13%]](http://slideplayer.com/slide/260973/1/images/44/I+%5BNote%3A+Approximately+13%25%5D.jpg "Definition Group A localized tumor completely removed with pathologically clear margins and no regional lymph node involvement. I [Note: Approximately 13%] A localized tumor that is grossly removed with: (A) microscopic disease at the margin, (B) involved, grossly removed regional lymph nodes, or (C) both A and B. II [Note: Approximately 20% ] A localized tumor with gross residual disease after incomplete removal or biopsy only. III [Note: Approximately 48% ] Distant metastases are present at diagnosis. IV [Note: Approximately 18% ]")
45
Group Stage Histology Risk Group I, II, III I, II 1 2, 3 Embryonal Low Risk III 1, 2, 3 Alveolar Intermediate Risk IV 4 Embryonal or Alveolar High Risk

46
Rhabdomyosarcoma - Treatment
Local control – Surgery vs. Radiation Systemic therapy – Chemotherapy Pediatric sarcomas are systemic illnesses

47
Rhabdomyosarcoma – Local Rx
Local control options: Surgery and radiation therapy The approach to local control of RMS depends on the site of origin RMS tends to occur is sites that are surgically challenging where attempts at radical resections may lead to mutilating surgery as well as inadequate surgical margins Use of radiation therapy is an important local control modality

48
Rhabdomyosarcoma – Surgery
Surgery in RMS is used with the aim of achieving complete resections with clear margins Potentially relevant disease sites: Vagina, paratesticular, non-parameningeal, non-orbit head & neck, extremity However – many children with RMS have tumors that cannot be excised or attempts at resection will lead to mutilation and loss of function (orbit, parameningeal, bladder) Consider radiaiton Late effects of radiation on young tissues
 Consider radiaiton. Late effects of radiation on young tissues.")
49
Rhabdomyosarcoma – Radiation Therapy
Required doses ~ Gy Essential in non –resectable cases and where surgical margins are inadequate (orbit, parameningeal, bladder) Tissue tolerance Late effects of radiation on young tissues
 Tissue tolerance. Late effects of radiation on young tissues.")
50
Rhabdomyosarcoma –Systemic Therapy
20% of patients present with metastatic disease Most patients (90%) who present without overt metastatic disease will develop systemic spread if not treated with chemotherapy (micro-metastatic disease) All patients must receive systemic therapy Active agents – Actinomycin, Cyclophosphamide/ifosfamide, vincristine, Doxorubicin, VP-16, topotecan/irinotecan
 who present without overt metastatic disease will develop systemic spread if not treated with chemotherapy (micro-metastatic disease) All patients must receive systemic therapy. Active agents – Actinomycin, Cyclophosphamide/ifosfamide, vincristine, Doxorubicin, VP-16, topotecan/irinotecan.")
51
Rhabdomyosarcoma – Treatment- COG
VCR / Actinomycin D / Cyclophosphamide 3 Wk wk wk Local Rx.(Surg/XRT) Cycle ↔ ↔ ↔ 4…………14………40 V V A A C C Vincristine – 2 mg/M2/course Actinomycin – 1.5 mg/M2/course Cyclophosphamide – 1200 mg/M2/course
 Cycle 1 ↔ 2 ↔ 3 ↔ 4…………14………40. V V. A A. C C. Vincristine – 2 mg/M2/course. Actinomycin – 1.5 mg/M2/course. Cyclophosphamide – 1200 mg/M2/course.")
52
Pediatric Cancer as a Systemic Illness – The rule and the exceptions
THE RULE- Pediatric solid tumors are always systemic – micrometastatic disease is present at diagnosis in the majority of patients All patients – including those with apparently localized disease - must be treated with chemotherapy Osteosarcoma, Ewing, RMS

53
Pediatric Cancer as a Systemic Illness – The rule and the exceptions
The exceptions: Tumors in which cure can be achieved with surgery alone Unilateral Retinoblastoma Stage I gonadal germ cell tumors Stage I-II hepatoblastoma Stage I – small – Wilms’ tumor Stage I neuroblastoma Supra-tentorial ependymomas Low grade gliomas

54
Rhabdomyosarcoma - Outcome
With the combination of local and systemic therapy – 50-70% of patients are cured Prognostic factors: Metastatic disease 10-20% (Lung > bone) Sites – favorable (orbit – 90%), unfavorable (extremity-60%) Histology: embryonal> alveolar
 Sites – favorable (orbit – 90%), unfavorable (extremity-60%) Histology: embryonal> alveolar.")
55
Rhabdomyosarcoma – Treatment of High Risk patients
Dose intensification – Alkylators Additional agents – doxorubicin, topotecan, irinotecan, ifosfamide, vinorelbine High dose chemotherapy with stem-cell rescue To date – none of these interventions have improved outcome

56
Rhabdomyosarcoma – Summary
RMS is the most common soft tissue sarcoma of childhood RMS can occur at multiple sites resulting in a wide spectrum of clinical presentations: The most common sites are 1) head and neck - including orbit and parameningeal, 2) genitourinary, including bladder, vagina and paratesticular 3) Extremities
 head and neck - including orbit and parameningeal, 2) genitourinary, including bladder, vagina and paratesticular 3) Extremities.")
57
Rhabdomyosarcoma – Summary -2
The diagnosis of RMS is made by a combination of clinical presentation, radiology and pathology The treatment of RMS is site specific Treatment of RMS must include a local component aimed at the primary tumor (surgery and/or radiation) and a systemic component (chemotherapy) aimed at micro-metastatic disease For most children with RMS a combination of vincristine, actinomycin and cyclophosphamide is the best current therapy
 and a systemic component (chemotherapy) aimed at micro-metastatic disease. For most children with RMS a combination of vincristine, actinomycin and cyclophosphamide is the best current therapy.")
58
Thank you

59
Rhabdomyosarcoma – Long term Sequelae
Site and treatment modality dependent Fertility – High doses of alkylating agents Cardiotoxicity – High doses of anthracyclines Second malignancies – AML (Topoisomerase+alkylators – 8-10%) Radiation field sarcomas (~5%)
 Radiation field sarcomas (~5%)")
60
Risk-Adapted Therapy Maximize benefit / Minimize risk
Patients with good-risk features and high cure rates – maintain good outcome, minimize toxicity (orbital, vaginal RMS) Patients with poor-risk features and low cure rates – intensify therapy (extremity and metastatic RMS), consider interventions to reduce long term toxicity
 Patients with poor-risk features and low cure rates – intensify therapy (extremity and metastatic RMS), consider interventions to reduce long term toxicity.")
Similar presentations




1 RETINOBLASTOMA. 2 RETINOBLASTOMA It is the most common primary ocular malignancy of childhood. It formed 15% of all childhood cancers.>")





.>")


>")








