Download presentation
Presentation is loading. Please wait.

1
1. Define, with examples, the following: a) Exon Splicing Enhancer b) Nonsense Mediated Decay c) Cryptic Splice Site How would you investigate the effect of a potential splicing mutation? Nicole Motton Birmingham RCPath Self-help Course Gene Structure & Function Session – 9th December 2008

2
Essay Plan Divided answer up in to headings by referring to the question: Introduction to splicing a)Describe exon splicing enhancer + give example(s) b)Describe nonsense mediated decay + give example(s) c)Describe cryptic splice site + give example(s) How would you investigate the effect of a potential splicing mutation? (Thought of work from a Sotos RNA case)
Describe exon splicing enhancer + give example(s) b)Describe nonsense mediated decay + give example(s) c)Describe cryptic splice site + give example(s) How would you investigate the effect of a potential splicing mutation (Thought of work from a Sotos RNA case)")
3
Introduction to splicing
RNA splicing removes non-essential RNA sequences from the primary transcript. Conserved sequences important for splicing include: Invariant GT and AG dinucleotides, located at the 5’ and 3’ ends of an intron respectively (the GT-AG rule). Rare AT-AC introns also exist. The immediately adjacent intronic and exonic sequences at the splice donor and splice acceptor sequences, including the polypyrimidine tract which precedes the end of an intron. The splice branch site (A is conserved in all genes). Splicing is known to be regulated through splice enhancer sequences and splice silencer sequences which occur within exons and introns.
. Rare AT-AC introns also exist. The immediately adjacent intronic and exonic sequences at the splice donor and splice acceptor sequences, including the polypyrimidine tract which precedes the end of an intron. The splice branch site (A is conserved in all genes). Splicing is known to be regulated through splice enhancer sequences and splice silencer sequences which occur within exons and introns.")
4
Spliceosome The splicing mechanism is mediated by a large RNA-protein complex, the spliceosome (made up of 5 small nuclear ribonucleoproteins (snRNPs) and more than 150 additional proteins). Acts in a processive manner: once a 5’ splice site is recognized, it scans the RNA sequence until it meets the next 3’ splice site (which would be signaled as a target by the branch site consensus sequence located just before it). Many genes naturally undergo alternative forms of RNA splicing. Mutations can sometimes produce an aberrant form of RNA splicing which is pathogenic.
 and more than 150 additional proteins). Acts in a processive manner: once a 5’ splice site is recognized, it scans the RNA sequence until it meets the next 3’ splice site (which would be signaled as a target by the branch site consensus sequence located just before it). Many genes naturally undergo alternative forms of RNA splicing. Mutations can sometimes produce an aberrant form of RNA splicing which is pathogenic.")
6
a) Exon Splicing Enhancer
Purine-rich regulatory sequences which can enhance splice site recognition by directing the splicing machinery to the appropriate sites and inhibit use of potential cryptic sites. Discrete but degenerate sequences of about 6-8 nucleotides long which are known to bind splice regulatory proteins. They are present in most if not all exons and a set of 10 ESE motifs have been predicted for human exons. Two major classes of ESEs have been defined based on nucleotide composition: purine-rich and A/C-rich. In mammalian cells the best characterized ESEs promote splicing by binding to the SR family of RNA-binding proteins (purine-rich) and recruit and stabilize binding of spliceosome components. The purine-rich ESEs are recognized by a conserved family of serine/arginine-rich (SR) proteins that recruit spliceosome components (such as U2 auxiliary factor, U2AF) to splice sites. ESEs can also enhance splicing by inhibiting adjacent ESSs. The A/C-rich ESEs (ACEs) bind the cold-box protein, YB-1, and promote splicing by an undetermined mechanism. SR family of RNA-binding proteins (have a distinctive C-terminal domain rich in serine-arginine dipeptides, a family of 11 highly conserved proteins that were originally identified as being required for constitutive and alternative splicing).
 and recruit and stabilize binding of spliceosome components. The purine-rich ESEs are recognized by a conserved family of serine/arginine-rich (SR) proteins that recruit spliceosome components (such as U2 auxiliary factor, U2AF) to splice sites. ESEs can also enhance splicing by inhibiting adjacent ESSs. The A/C-rich ESEs (ACEs) bind the cold-box protein, YB-1, and promote splicing by an undetermined mechanism. SR family of RNA-binding proteins (have a distinctive C-terminal domain rich in serine-arginine dipeptides, a family of 11 highly conserved proteins that were originally identified as being required for constitutive and alternative splicing).")
8
ESE Example 1 SMN2 exon 7 base substitution (g.C6T) was thought to disrupt an ESE in exon 7 that causes the exon to be skipped in the majority of SMN2 mRNAs. The resulting SMN2 delE7 mRNA encodes a truncated protein missing the C-terminal 16 residues and is thought to be nonfunctional. But, more recent paper contests this saying SMN2 exon 7 splicing is repressed by creation of an hnRNPA1-dependent ESS. The best characterized ESSs and ISSs repress splicing by binding to heterogeneous nuclear ribonucleoproteins (hnRNPs are an abundant class of predominantly nuclear RNA binding proteins that contain an RNA recognition motif type of RNA binding domain). These proteins promote various steps in assembly of spliceosomes and also bind to splicing enhancer sequences.
 was thought to disrupt an ESE in exon 7 that causes the exon to be skipped in the majority of SMN2 mRNAs. The resulting SMN2 delE7 mRNA encodes a truncated protein missing the C-terminal 16 residues and is thought to be nonfunctional. But, more recent paper contests this saying SMN2 exon 7 splicing is repressed by creation of an hnRNPA1-dependent ESS. The best characterized ESSs and ISSs repress splicing by binding to heterogeneous nuclear ribonucleoproteins (hnRNPs are an abundant class of predominantly nuclear RNA binding proteins that contain an RNA recognition motif type of RNA binding domain). These proteins promote various steps in assembly of spliceosomes and also bind to splicing enhancer sequences.")
9
ESE Example 2 A missense mutation in exon 5 of the medium-chain acyl-CoA dehydrogenase gene (MCAD) inactivates an ESE in exon 5 causing exon skipping and MCAD deficiency, resulting in a disease that affects mitochondrial function. Interestingly, a silent polymorphism in the same exon (11 nucleotides upstream) of the mutation modulates the severity of exon skipping by inactivating an adjacent ESS. This is an example of a cis-acting modifier and illustrates the relationship between disease-causing splicing mutations and disease-modifying natural variants that affect splicing.
 inactivates an ESE in exon 5 causing exon skipping and MCAD deficiency, resulting in a disease that affects mitochondrial function. Interestingly, a silent polymorphism in the same exon (11 nucleotides upstream) of the mutation modulates the severity of exon skipping by inactivating an adjacent ESS. This is an example of a cis-acting modifier and illustrates the relationship between disease-causing splicing mutations and disease-modifying natural variants that affect splicing.")
10
ESE Example 3 3 bp deletion and several point mutations (silent, missense, nonsense) disrupt an exon splicing enhancer in exon 3 of MLH1 and is the cause of HNPCC in a Quebec family. All the mutations cause varying degrees of exon skipping, suggesting the presence of an ESE at the 5' end of exon 3. These mutations are situated in a GAAGAT sequence 3 bp downstream from the start of exon 3.
 disrupt an exon splicing enhancer in exon 3 of MLH1 and is the cause of HNPCC in a Quebec family. All the mutations cause varying degrees of exon skipping, suggesting the presence of an ESE at the 5 end of exon 3. These mutations are situated in a GAAGAT sequence 3 bp downstream from the start of exon 3.")
11
b)Nonsense Mediated Decay
This is a form of in vivo RNA surveillance that is believed to have evolved to protect the body from the possible consequences of truncated proteins interfering with normal function. Nonsense-mediated mRNA decay (NMD) largely functions to ensure the quality of gene expression. NMD is also crucial to regulating appropriate expression levels for certain genes and for maintaining genome stability. NMD is a means to degrade abnormal mRNAs that prematurely terminate translation (due to frameshift or nonsense mutations). NMD targets can also arise as a result of error in cellular processes (inefficiently spliced pre-mRNAs, transcripts from T-cell receptor genes, immunoglobulin genes and other antigen-receptor genes that undergo error prone somatic-cell DNA rearrangements and hypermutations during lymphocyte development). NMD and the proteins of the NMD pathway have other functions other than mRNA quality control: regulation of the expression of certain classes of genes, roles in specialized pathways of mRNA decay, in stress-response pathways and DNA repair, functions in DNA synthesis and cell-cycle progression, and the maintenance of telomeres.
 largely functions to ensure the quality of gene expression. NMD is also crucial to regulating appropriate expression levels for certain genes and for maintaining genome stability. NMD is a means to degrade abnormal mRNAs that prematurely terminate translation (due to frameshift or nonsense mutations). NMD targets can also arise as a result of error in cellular processes (inefficiently spliced pre-mRNAs, transcripts from T-cell receptor genes, immunoglobulin genes and other antigen-receptor genes that undergo error prone somatic-cell DNA rearrangements and hypermutations during lymphocyte development). NMD and the proteins of the NMD pathway have other functions other than mRNA quality control: regulation of the expression of certain classes of genes, roles in specialized pathways of mRNA decay, in stress-response pathways and DNA repair, functions in DNA synthesis and cell-cycle progression, and the maintenance of telomeres.")
12
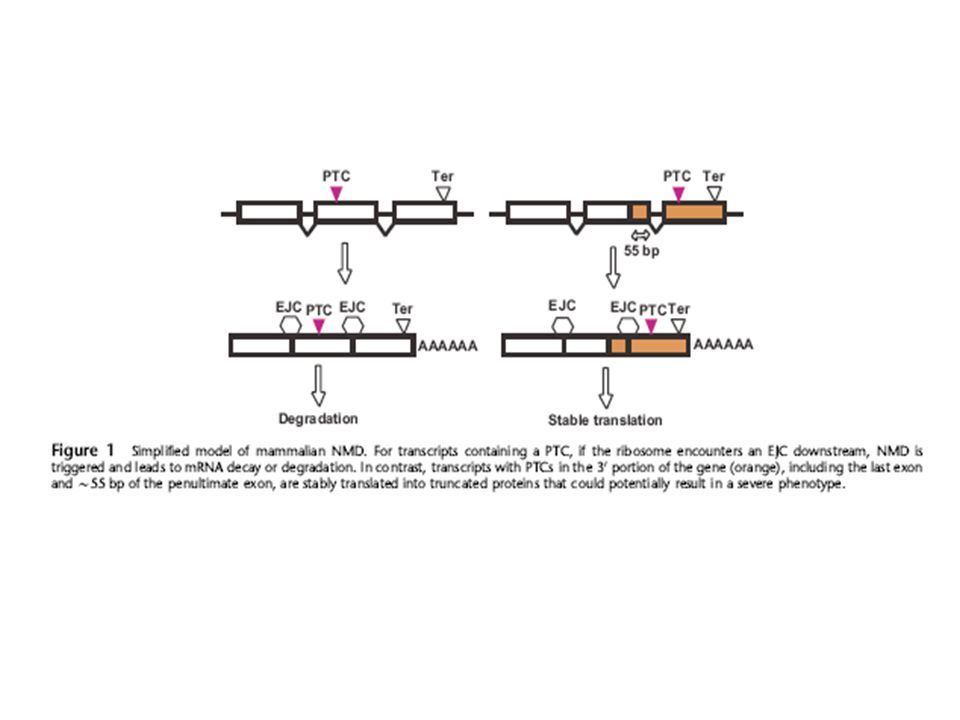
Mutations can introduce premature termination codons.
NMD is usually triggered when translation terminates prematurely at one of the three nonsense codons i.e. premature termination codon (PTCs). Mutations can introduce premature termination codons. In mammalian NMD, an intron apparently functions as a second signal for triggering NMD by leaving a ‘mark’ on the mRNA at the exon-exon junction as a consequence of the splicing event. This mark known as the exon-junction complex (EJC) enables the NMD RNA surveillance pathway to differentiate between PTCs and normal stop codons present in the last exon. The EJC is a complex of proteins that is deposited ~20-25 nucleotides upstream of the exon-exon junctions of newly synthesized spliced mRNAs. The mammalian NMD surveillance system cannot distinguish PTCs in the penultimate exon that are located <~55nt from the final intron. (A translocating ribosome would displace an EJC upstream of the last exon-exon junction before the ribosome could recognize a stop codon located <55nt from the exon-exon junction.) Mutations that introduce PTCs e.g. nonsense mutations by direct substitution of normal codon to a stop codon, frameshift insertion or deletion mutations introduce a premature termination codon downstream of mutation site, splice-site mutations can introduce a premature termination codon (e.g by skipping an exon containing a number of nucleotides that cannot be divided by three). The inability to differentiate nonsense codon mutations in the last and 3’ end of the penultimate exon from the normal termination codon can result in stable translation of mRNAs that contain PTCs located within these ‘protected’ regions. Such an escape from NMD surveillance may cause expression of large amounts of aberrant truncated proteins with potential dominant-negative or gain-of-function (particularly relevant to disease genes with large last exons) effects in cells.
. Mutations can introduce premature termination codons. In mammalian NMD, an intron apparently functions as a second signal for triggering NMD by leaving a ‘mark’ on the mRNA at the exon-exon junction as a consequence of the splicing event. This mark known as the exon-junction complex (EJC) enables the NMD RNA surveillance pathway to differentiate between PTCs and normal stop codons present in the last exon. The EJC is a complex of proteins that is deposited ~20-25 nucleotides upstream of the exon-exon junctions of newly synthesized spliced mRNAs. The mammalian NMD surveillance system cannot distinguish PTCs in the penultimate exon that are located <~55nt from the final intron. (A translocating ribosome would displace an EJC upstream of the last exon-exon junction before the ribosome could recognize a stop codon located <55nt from the exon-exon junction.) Mutations that introduce PTCs e.g. nonsense mutations by direct substitution of normal codon to a stop codon, frameshift insertion or deletion mutations introduce a premature termination codon downstream of mutation site, splice-site mutations can introduce a premature termination codon (e.g by skipping an exon containing a number of nucleotides that cannot be divided by three). The inability to differentiate nonsense codon mutations in the last and 3’ end of the penultimate exon from the normal termination codon can result in stable translation of mRNAs that contain PTCs located within these ‘protected’ regions. Such an escape from NMD surveillance may cause expression of large amounts of aberrant truncated proteins with potential dominant-negative or gain-of-function (particularly relevant to disease genes with large last exons) effects in cells.")
14
Two central NMD-pathway proteins are:
up-frameshift 1 (UPF) phosphatidylinositol 3-kinase-related protein (PIKK) (SMG) these are important in a number of cellular pathways other than NMD. It is unclear how cells direct NMD factors to one or another function. Protein function can be influenced by binding partners; UPF1 binds to an EJC in NMD. NMD can effect clinical outcome in at least three major ways (1) altering the pattern of inheritance (2) causing distinct traits to manifest from mutations in the same gene (3) modifying the specific clinical phenotype NMD has a role as a modifier of the phenotypic consequences of PTC. Ultimately the phenotypic outcome depends on the function of mutant protein and escape from NMD does not necessarily result in a more severe phenotype. Many genetic diseases that were previously thought to be the result of dominant-negative mutations may actually result from loss-of-function or haploinsufficiency in vivo because of NMD.
 phosphatidylinositol 3-kinase-related protein (PIKK) (SMG) these are important in a number of cellular pathways other than NMD. It is unclear how cells direct NMD factors to one or another function. Protein function can be influenced by binding partners; UPF1 binds to an EJC in NMD. NMD can effect clinical outcome in at least three major ways. (1) altering the pattern of inheritance. (2) causing distinct traits to manifest from mutations in the same gene. (3) modifying the specific clinical phenotype. NMD has a role as a modifier of the phenotypic consequences of PTC. Ultimately the phenotypic outcome depends on the function of mutant protein and escape from NMD does not necessarily result in a more severe phenotype. Many genetic diseases that were previously thought to be the result of dominant-negative mutations may actually result from loss-of-function or haploinsufficiency in vivo because of NMD.")
15
Models for nonsense-mediated mRNA decay in Saccharomyces cerevisiae & mammals.
a | In Saccharomyces cerevisiae, newly synthesized mRNAs that contain a premature termination codon (PTC) and that are bound to the RNA cap-binding protein heterodimer Cbc1–Cbc2 and to steady-state mRNAs that are bound by the cap-binding protein eukaryotic translation initiation factor 4E (eIF4E) are targeted for nonsense-mediated mRNA decay (NMD) once the mRNA is exported from the nucleus to the cytoplasm. In at least one mechanism, an abnormally long or 'faux' 3' UTR results in inefficient translation termination. As a consequence, termination involves not only the eukaryotic release factor 1 (eRF1) and eRF3 translation-termination factors, which fail to effectively mediate the release of the nascent polypeptide because of an inefficient interaction between eRF3 and poly(A)-binding protein 1 (Pab1), but probably also the up-frameshift 1 (Upf1), Upf2 and Upf3 NMD factors. These factors then recruit and/or activate mRNA degradative activities. Although Upf1 is a phosphoprotein, whether Upf1 undergoes a cycle of phosphorylation and dephosphorylation during NMD in S. cerevisiae is unknown. b | In mammals, newly synthesized PTC-containing mRNA that is bound to the RNA cap-binding protein heterodimer CPB80–CBP20 is targeted for NMD once the mRNA has been generated by pre-mRNA splicing and exported from the nucleus to the cytoplasm. Notably, pre-mRNA splicing results in the deposition of an exon-junction complex (EJC) of proteins upstream of mRNA exon–exon junctions. Core EJC components consist of eIF4AIII, RNA-binding-motif protein Y14, mago nashi homologue (MAGOH) and Barentsz (BTZ; also known as cancer susceptibility candidate 3, CASC3). The UPF3 or UPF3X NMD factor, which shuttles to the nucleus, is thought to be recruited to EJCs in the nucleus and is exported with the mRNA to the cytoplasm. UPF3 or UPF3X then recruits UPF2, which is primarily cytoplasmic. The translation of mRNA that is bound to CBP80–CBP20 is termed the pioneer round. Translation termination at a PTC during the pioneer round involves the SURF complex, which consists of the phosphatidylinositol 3-kinase-related protein kinase (PIKK) SMG1 together with UPF1, eRF1 and eRF3. Generally, if translation terminates more than 50–55 nucleotides (nt) upstream of an exon–exon junction (that is, more than 25–30 nt upstream of an EJC), then NMD will occur. UPF1, together with SMG1, is thought to bind EJC-associated UPF2 in a way that is promoted by CBP80 (not shown). UPF1 binding to the EJC triggers UPF1 phosphorylation and NMD by promoting translational repression and recruiting mRNA degradative activities. Not shown are SMG5, SMG6 and SMG7, which seem to recruit protein phosphatase 2A (PP2A) and function in UPF1 dephosphorylation and, thus, recycling. AUG, translation initiation codon; STOP, normal termination codon. Olaf Isken & Lynne E. Maquat. Nature Reviews Genetics 9, (September 2008)
 and that are bound to the RNA cap-binding protein heterodimer Cbc1–Cbc2 and to steady-state mRNAs that are bound by the cap-binding protein eukaryotic translation initiation factor 4E (eIF4E) are targeted for nonsense-mediated mRNA decay (NMD) once the mRNA is exported from the nucleus to the cytoplasm. In at least one mechanism, an abnormally long or faux 3 UTR results in inefficient translation termination. As a consequence, termination involves not only the eukaryotic release factor 1 (eRF1) and eRF3 translation-termination factors, which fail to effectively mediate the release of the nascent polypeptide because of an inefficient interaction between eRF3 and poly(A)-binding protein 1 (Pab1), but probably also the up-frameshift 1 (Upf1), Upf2 and Upf3 NMD factors. These factors then recruit and/or activate mRNA degradative activities. Although Upf1 is a phosphoprotein, whether Upf1 undergoes a cycle of phosphorylation and dephosphorylation during NMD in S. cerevisiae is unknown. b | In mammals, newly synthesized PTC-containing mRNA that is bound to the RNA cap-binding protein heterodimer CPB80–CBP20 is targeted for NMD once the mRNA has been generated by pre-mRNA splicing and exported from the nucleus to the cytoplasm. Notably, pre-mRNA splicing results in the deposition of an exon-junction complex (EJC) of proteins upstream of mRNA exon–exon junctions. Core EJC components consist of eIF4AIII, RNA-binding-motif protein Y14, mago nashi homologue (MAGOH) and Barentsz (BTZ; also known as cancer susceptibility candidate 3, CASC3). The UPF3 or UPF3X NMD factor, which shuttles to the nucleus, is thought to be recruited to EJCs in the nucleus and is exported with the mRNA to the cytoplasm. UPF3 or UPF3X then recruits UPF2, which is primarily cytoplasmic. The translation of mRNA that is bound to CBP80–CBP20 is termed the pioneer round. Translation termination at a PTC during the pioneer round involves the SURF complex, which consists of the phosphatidylinositol 3-kinase-related protein kinase (PIKK) SMG1 together with UPF1, eRF1 and eRF3. Generally, if translation terminates more than 50–55 nucleotides (nt) upstream of an exon–exon junction (that is, more than 25–30 nt upstream of an EJC), then NMD will occur. UPF1, together with SMG1, is thought to bind EJC-associated UPF2 in a way that is promoted by CBP80 (not shown). UPF1 binding to the EJC triggers UPF1 phosphorylation and NMD by promoting translational repression and recruiting mRNA degradative activities. Not shown are SMG5, SMG6 and SMG7, which seem to recruit protein phosphatase 2A (PP2A) and function in UPF1 dephosphorylation and, thus, recycling. AUG, translation initiation codon; STOP, normal termination codon. Olaf Isken & Lynne E. Maquat. Nature Reviews Genetics 9, (September 2008)")
16
UPF1 and SMG1 are at the interface of processes important for gene and genome regulation in mammalian cells. (Anticlockwise from the upper left). Up-frameshift 1 (UPF1) and phosphatidylinositol 3-kinase-related protein kinase SMG1 are involved in multiple RNA and DNA surveillance pathways. UPF1 functions during nonsense-mediated mRNA decay (NMD), in which translation termination that occurs sufficiently upstream of a splicing-generated exon-junction complex (EJC), usually at a premature termination codon (PTC), results in SMG1-mediated UPF1 phosphorylation and, as a consequence, mRNA decay. UPF1 is also instrumental to the RNA-binding protein Staufen 1 (STAU1)-mediated mRNA decay (SMD) and replication-dependent histone mRNA decay, during which it is recruited to specific 3' UTRs by STAU1 (in SMD) or stem-loop binding protein (SLBP) (in replication-dependent histone mRNA decay). Although the UPF1 kinase involved in SMD is unknown, ataxia-telangiectasia mutated and Rad 3-related (ATR), ataxia-telangiectasia mutated (ATM) and DNA-dependent protein kinase (DNA-PK) seem to phosphorylate UPF1 during histone mRNA decay. Additionally, UPF1 associates with DNA polymerase (Pol) and is essential for human cells to complete DNA replication and repair in a process that involves ATR if not other phosphatidylinositol 3-kinase-related protein kinases (PIKKs). Finally, UPF and SMG proteins seem to function in genome stability to modulate telomerase function and to regulate telomere length: they are enriched at telomeres so as to negatively regulate telomeric repeat-containing RNA (TERRA) association with telomeric chromosomes. AUG, translation initiation codon; BTZ, Barentsz (also known as cancer susceptibility candidate 3, CASC3); eIF4AIII, eukaryotic initiation factor 4AIII; MAGOH, mago nashi homologue; PTC, premature translation termination codon; SBS, STAU1 binding site; STOP, normal termination codon; Y14, RNA-binding motif-protein Y14. Olaf Isken & Lynne E. Maquat. Nature Reviews Genetics 9, (September 2008)
. Up-frameshift 1 (UPF1) and phosphatidylinositol 3-kinase-related protein kinase SMG1 are involved in multiple RNA and DNA surveillance pathways. UPF1 functions during nonsense-mediated mRNA decay (NMD), in which translation termination that occurs sufficiently upstream of a splicing-generated exon-junction complex (EJC), usually at a premature termination codon (PTC), results in SMG1-mediated UPF1 phosphorylation and, as a consequence, mRNA decay. UPF1 is also instrumental to the RNA-binding protein Staufen 1 (STAU1)-mediated mRNA decay (SMD) and replication-dependent histone mRNA decay, during which it is recruited to specific 3 UTRs by STAU1 (in SMD) or stem-loop binding protein (SLBP) (in replication-dependent histone mRNA decay). Although the UPF1 kinase involved in SMD is unknown, ataxia-telangiectasia mutated and Rad 3-related (ATR), ataxia-telangiectasia mutated (ATM) and DNA-dependent protein kinase (DNA-PK) seem to phosphorylate UPF1 during histone mRNA decay. Additionally, UPF1 associates with DNA polymerase (Pol) and is essential for human cells to complete DNA replication and repair in a process that involves ATR if not other phosphatidylinositol 3-kinase-related protein kinases (PIKKs). Finally, UPF and SMG proteins seem to function in genome stability to modulate telomerase function and to regulate telomere length: they are enriched at telomeres so as to negatively regulate telomeric repeat-containing RNA (TERRA) association with telomeric chromosomes. AUG, translation initiation codon; BTZ, Barentsz (also known as cancer susceptibility candidate 3, CASC3); eIF4AIII, eukaryotic initiation factor 4AIII; MAGOH, mago nashi homologue; PTC, premature translation termination codon; SBS, STAU1 binding site; STOP, normal termination codon; Y14, RNA-binding motif-protein Y14. Olaf Isken & Lynne E. Maquat. Nature Reviews Genetics 9, (September 2008)")
17
NMD Example 1 β-Globin p.Gln39X
The occurrence of both dominant and recessive mutant alleles in a single gene due to NMD. β-thalassaemia represents an example in which 5’ PTCs in the β-globin gene result in a recessive trait, whereas 3’ PTCs can result in an atypical dominant form of disease. 5’ PTCs but not 3’ PTCs trigger β-globin NMD. The decrease/absent mutant mRNA levels cause a recessive mode of inheritance.

18
NMD Example 2 Rare truncating mutations that occur near the 3’ end of the dystrophin gene result in variable mild phenotypes, suggesting these truncating proteins are capable of a partial rescue of the DMD phenotype. The 5’ PTCs are associated with a severe form of DMD and fail to rescue the phenotype because of NMD.

19
c) Cryptic Splice Site Cryptic (or latent) splice sites coincidentally resemble the sequences of authentic splice sites but are not normally used in splicing unless (1) a mutation directly activates a cryptic splice site by changing its sequence so that it becomes more like the consensus splice donor or acceptor sequence. The altered cryptic splice site can now be recognized and used by the spliceosome (direct activation) (2) a mutation occurs in an authentic splice site, causing a splice donor or splice acceptor to be faulty, in which case the splicing apparatus scans for other possible alternatives and selects a cryptic splice site (indirect activation) Because individual splice donor and splice acceptor sequences often show some variation from the consensus sequences, cryptic splice sites occur frequently within genes.
 splice sites coincidentally resemble the sequences of authentic splice sites but are not normally used in splicing unless. (1) a mutation directly activates a cryptic splice site by changing its sequence so that it becomes more like the consensus splice donor or acceptor sequence. The altered cryptic splice site can now be recognized and used by the spliceosome (direct activation) (2) a mutation occurs in an authentic splice site, causing a splice donor or splice acceptor to be faulty, in which case the splicing apparatus scans for other possible alternatives and selects a cryptic splice site (indirect activation) Because individual splice donor and splice acceptor sequences often show some variation from the consensus sequences, cryptic splice sites occur frequently within genes.")
20
Exon skipping may occur if an alternative splice site is used.
Mutation of a splice donor sequence often results in skipping of the upstream exon. Mutation of the splice acceptor sequence often results in skipping of the downstream exon. However, use of an alternative exonic or intronic cryptic splice site means the outcome is not always easily predictable. When an exon is skipped, various outcomes are possible. If the number of nucleotides in the exon is not divisible by three, a frameshift will introduce a premature termination codon, often resulting in an unstable RNA transcript and no polypeptide. If exon skipping does not cause a frameshift, the absence of the normally encoded amino acids will often result in a nonfunctional or abnormal polypeptide depending on the importance of these amino acids to protein function and/or structure.

21
Cryptic Splice Site Example 1
Examples of activation of a cryptic splice acceptor within an intron and a cryptic splice donor site within an exon (apparently silent mutations may yet be pathogenic). Activate a cryptic splice site: CFTR c kbC>T point mutation in intron 19 creates a variably spliced 84-nucleotide exon (activates a cryptic splice site deep within an intron). Variable level of cryptic exon inclusion influences disease severity.
. Activate a cryptic splice site: CFTR c kbC>T point mutation in intron 19 creates a variably spliced 84-nucleotide exon (activates a cryptic splice site deep within an intron). Variable level of cryptic exon inclusion influences disease severity.")
22
Cryptic Splice Site Example 2
Silent mutation (p.Gly624Gly) in a LGMD2A limb girdle muscular dystrophy patient. The mutation was found in the calpain 3 gene at the third base of a codon and appeared to be a silent mutation. Leads to replacement of one glycine codon (GGC) by another glycine codon (GGT). However, the mutation is believed to be pathogenic because the substitution results in activation of a cryptic splice donor sequence (AGGGCAAAAG) within exon 16 resulting in aberrant splicing with the loss of coding sequence from exon 16 and the introduction of a frameshift.
 in a LGMD2A limb girdle muscular dystrophy patient. The mutation was found in the calpain 3 gene at the third base of a codon and appeared to be a silent mutation. Leads to replacement of one glycine codon (GGC) by another glycine codon (GGT). However, the mutation is believed to be pathogenic because the substitution results in activation of a cryptic splice donor sequence (AGGGCAAAAG) within exon 16 resulting in aberrant splicing with the loss of coding sequence from exon 16 and the introduction of a frameshift.")
23
How would you investigate the effect of a potential splicing mutation?
Mutations that involve the invariant GT-AG splice donor and acceptor sites assume pathogenic. So investigations are of mutations in the intron (which may introduce a cryptic splice-site, disrupt the branch site) or in the exon (which may disrupt an exonic splice enhancer or introduce a cryptic splice).
 or in the exon (which may disrupt an exonic splice enhancer or introduce a cryptic splice).")
24
Functional studies – protein-based functional assay to divide the product in to functional or non-functional. They are specific to a particular protein. Protein expression - e.g. HNPCC - IHC analysis to look at expression of the proteins of the mismatch repair genes e.g. MLH1, MSH2, MSH6, PMS2. RNA studies - RT-PCR can be used to detect aberrant splicing. Problem with NMD in that the RT-PCR product from a heterozygous person may show only the normal allele. Mutations deep within the intron may be overlooked unless studied using RT-PCR e.g. the CFTR c kbC>T activates a cryptic splice site that lies 10kb inside intron 19. Databases and literature – has it been seen in disease patients before?

25
Screen normal controls (100)
Family studies – de novo mutation not present in parents, in a person with a de novo disease (Sotos – mainly de novo with rare familial cases). For cancer is the change present in other affected relatives? In silico – the nature of the sequence change. Amino acid substitutions are more likely to affect function if they are nonconservative (replace a polar by a nonpolar amino acid, or an acidic by a basic one). Is it in a functionally important domain? Is the amino acid conserved in related genes? Species conservation? ESE motif-prediction tools. Splice-site prediction tools like fruitfly. SIFT (Sorting Intolerant From Tolerant is a program that predicts whether an amino acid substitution affects protein function so that users can prioritize substitutions for further study) Polyphen (Polymorphism Phenotyping is a tool which predicts possible impact of an amino acid substitution on the structure and function of a human protein using straightforward physical and comparative considerations). Screen normal controls (100)
. For cancer is the change present in other affected relatives In silico – the nature of the sequence change. Amino acid substitutions are more likely to affect function if they are nonconservative (replace a polar by a nonpolar amino acid, or an acidic by a basic one). Is it in a functionally important domain Is the amino acid conserved in related genes Species conservation ESE motif-prediction tools. Splice-site prediction tools like fruitfly. SIFT (Sorting Intolerant From Tolerant is a program that predicts whether an amino acid substitution affects protein function so that users can prioritize substitutions for further study) Polyphen (Polymorphism Phenotyping is a tool which predicts possible impact of an amino acid substitution on the structure and function of a human protein using straightforward physical and comparative considerations). Screen normal controls (100)")
26
Minigenes A minigene contains a genomic fragment including the alternative exon(s) and the surrounding introns as well as the flanking constitutively spliced cloned in a eukaryotic expression vector. Thus, the transfected minigenes should contain all RNA-elements necessary to show the same alternative splicing pattern as the corresponding endogenous alternatively spliced gene when compared in a specific cellular environment. Splicing microarrays are used for large scale identification of splicing differences between two RNA populations. Predicted splicing events validated by RT-PCR.
 and the surrounding introns as well as the flanking constitutively spliced cloned in a eukaryotic expression vector. Thus, the transfected minigenes should contain all RNA-elements necessary to show the same alternative splicing pattern as the corresponding endogenous alternatively spliced gene when compared in a specific cellular environment. Splicing microarrays are used for large scale identification of splicing differences between two RNA populations. Predicted splicing events validated by RT-PCR.")
27
Splicing Microarray

28
Sotos case: disruption of branch site.
Sequencing analysis identified a novel variant in intron 21 of the NSD1 gene (c T>G) of unknown clinical significance. Parental DNA samples were provided and showed no evidence of this variant. Given the location of this variant within the lariat branch site it was predicted to interfere with the normal splicing process.
 of unknown clinical significance. Parental DNA samples were provided and showed no evidence of this variant. Given the location of this variant within the lariat branch site it was predicted to interfere with the normal splicing process.")
29
mRNA was extracted from a fresh blood sample from the patient.
RT-PCR followed by sequence analysis using exonic primers flanking exon 22 was used to investigate the effect of an intronic variant in intron 21. A larger aberrant product was seen in the patient cDNA at a similar level to the wild type. Analysis of mRNA from the patient showed the inclusion of 30bp of intron 21 of the NSD1 gene (r.[ _6259-1ins; u>g]+[=]) that directly resulted in the introduction of a premature termination codon (p.[Asn2087_Gln2088insAlaAspCysSerIleSerAspLeuX]+[=]). It is assumed that this is a direct consequence of the variant disrupting the consensus sequence of the branch site upstream from the acceptor site of exon 22.
 that directly resulted in the introduction of a premature termination codon (p.[Asn2087_Gln2088insAlaAspCysSerIleSerAspLeuX]+[=]). It is assumed that this is a direct consequence of the variant disrupting the consensus sequence of the branch site upstream from the acceptor site of exon 22.")
30
References Strachan and Read, Human Molecular Genetics 3.
Splicing in disease: disruption of the splicing code and the decoding machinery. Guey-Shin Wang and Thomas A Cooper. Nature Reviews Genetics, Vol. 8, October 2007: Pre-mRNA splicing and human disease. Nuno Andre Faustino and Thomas A Cooper. Genes and Development (2008) 17: hnRNP A1 functions with specificity in repression of SMN2 exon 7 splicing. Kashima et al. Human Molecular Genetics (2007) Vol 16, No 24: Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Khajavi et al. European Journal of Human Genetics (2006) Vol 14: The multiple lives of NMD factors: balancing roles in gene and genome regulation. Olaf Isken and Lynne E. Maquat. Nature Reviews Genetics, Vol 9, September 2008: Kashima et al, Human Molecular Genetics, 2007, Vol 16, no24: McVety et al. J Med Genet.2006; 43:
 17: hnRNP A1 functions with specificity in repression of SMN2 exon 7 splicing. Kashima et al. Human Molecular Genetics (2007) Vol 16, No 24: Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Khajavi et al. European Journal of Human Genetics (2006) Vol 14: The multiple lives of NMD factors: balancing roles in gene and genome regulation. Olaf Isken and Lynne E. Maquat. Nature Reviews Genetics, Vol 9, September 2008: Kashima et al, Human Molecular Genetics, 2007, Vol 16, no24: McVety et al. J Med Genet.2006; 43:")
Similar presentations




>")




 Researchers use the following techniques to find DNA sequences involved in regulation: – Deletion mapping – DNA footprinting.>")










 Umm-e-Habiba(BB307-035). Gene splicing “Gene splicing is the removal of introns from the primary trascript of a discontinuous gene.>")
