Download presentation
Presentation is loading. Please wait.

1
Introduction to Amber The theory and practice of biomolecular simulations using the Amber suite of programs Dr. Vladislav Vassiliev NCI National Facility, The Australian National University, ACT 0200, Canberra, Australia February 2011 1

2
Presentation Outline Introduction to Amber 12 Hands-on
Setting up a standard Amber MD Run Building non-standard Residues QM/MM: Using Amber-Gaussian Interface QM/MM: Using Amber inbuilt QM methods 2

3
What is AMBER? Assisted Model Building with Energy Refinement AMBER 3

4
What is Amber? “Amber” refers to two things:
1) a set of molecular mechanical force fields for the simulation of biomolecules 2) a package of molecular simulation programs (about 50 ) which includes source code and demos The current version of the code is Amber version 12, which is distributed by UCSF (University of California, San Francisco) subject to a licensing agreement Amber Home Page:
 a set of molecular mechanical force fields for the simulation of biomolecules. 2) a package of molecular simulation programs (about 50 ) which includes source code and demos. The current version of the code is Amber version 12, which is distributed by UCSF (University of California, San Francisco) subject to a licensing agreement. Amber Home Page:")
5
What is Amber Amber is distributed in two parts:
AmberTools12 and Amber 12: AmberTools12 could be used without Amber12, but not vice versa AmberTools12 currently consists of several independently developed packages that work well by themselves, and with Amber itself Amber 12 centered around the sander and pmemd simulation programs and continues to be licensed as before, under a more restrictive license (Academic/non-profit/government: $400. Industrial (for-profit): $20,000 for new licensees, $15,000 for licensees of Amber 10).
: $20,000 for new licensees, $15,000 for licensees of Amber 10).")
6
AmberTools NAB antechamber & MCPB ptraj & cpptraj tleap and xleap
build molecules; run MD or distance geometry, etc. antechamber & MCPB Create force fields for general organic molecules ptraj & cpptraj Analyze trajectories from Amber or CHARMM tleap and xleap Basic preparation program for Amber simulations 3D-RISM Solves integral equation models for solvation sqm semiempirical and DFTB quantum chemistry pbsa Performs numerical solutions to Poisson-Boltzmann models Mdgx Code for explicit solvent molecular dynamics simulations MMPBSA.py & amberlite Energy-based analyses of MD trajectories

7
AmberTools AmberTools is released under the GNU General Public License (GPL) A few components are included that are in the public domain or which have other, open-source, licenses. AmberTools is distributed in source code format, and must be compiled in order to be used. One needs C, C++, and fortran compilers to compile the AmberTools programs. The source code of AmberTools could be obtained here:

8
Versions of Amber Version Released 12 2012 11 2010 10 2008 9 2006 8
2004 8

9
AMBER Home Have a look at the Amber Home Page: 9

10
Amber Main References A general overview of the Amber codes:
D.A. Case, T.E. Cheatham, III, T. Darden, H. Gohlke, R. Luo, K.M. Merz, Jr., A. Onufriev, C. Simmerling, B. Wang and R. Woods. The Amber biomolecular simulation programs. J. Comput. Chem. 26, (2005) An overview of the Amber protein force fields, and how they were developed: W. Ponder and D.A. Case. Force fields for protein simulations. Adv. Prot. Chem. 66, (2003). E. Cheatham, III and M.A. Young. Molecular dynamics simulation of nucleic acids: Successes, limitations and promise. Biopolymers 56, (2001). 10
 An overview of the Amber protein force fields, and how they were developed: W. Ponder and D.A. Case. Force fields for protein simulations. Adv. Prot. Chem. 66, (2003). E. Cheatham, III and M.A. Young. Molecular dynamics simulation of nucleic acids: Successes, limitations and promise. Biopolymers 56, (2001). 10.")
11
“Amber” is a software package for modelling of Large Molecular Systems
What is Amber? “Amber” is a software package for modelling of Large Molecular Systems 11

12
Why Do We Need A Special Treatment for Large Molecular Systems?
Or, Why Do We Need Amber? 12

13
Quantum Chemistry Methods Provide a Rigorous Description of Molecular Systems
They solve Schrödinger equation And they are generally applicable: But… they are very time consuming… 13

14
To Treat Large Molecular Systems We Need to Reduce the Complexity of the System
Molecular Mechanics is a non-quantum mechanical technique for treating Large Molecular Systems As a result Molecular Mechanics methods are thousands times faster than Quantum Chemistry methods 14

15
Molecular Mechanics vs Quantum Mechanics
Considers atoms as collections of electrons and nuclei Considers atoms as soft or hard spheres. Covalent bonds are treated as springs Solves quantum Schrödinger equation Uses classical potential energy equations 15

16
Force Fields The potential energy equations to calculate the energy in Molecular Mechanics methods and the parameters/constants used in the equations are known as a Force Field There are many force fields designed for different purposes. QVBMM SIBFA UFF MM2, MM3, MM4 COSMOS-NMR AMBER DRF90 PIPF OPLS MMFF ECEPP/2 CFF ENZYMIX GROMACS CHARMm X-Pol CVFF QCFF/PI GROMOS AMOEBA CHARMM 16

17
Etotal = Ebonded + Enonbonded
Amber Force Field The total Energy in Amber force field consists of bonded terms relating to atoms linked by covalent bonds and nonbonded terms describing the long-range electrostatic and van der Waals interactions: Etotal = Ebonded + Enonbonded 17

18
Amber Force Field: Bonded Terms Ebonded = Estretch + Ebend + Etorsion
The bonded Energy in Amber force field consists of bond stretching, angle bending, and torsion terms: Ebonded = Estretch + Ebend + Etorsion 18

19
Amber Force Field: Bond Stretching
Amber force field treats covalent bonds between atoms as springs (Hooke's Law, F = -kx) where Kr is the empirical stretching force constant, r is the actual bond length and req is the “natural” (empirical) bond length 19
 where Kr is the empirical stretching force constant, r is the actual bond length and req is the natural (empirical) bond length. 19.")
20
Amber Force Field: Bond Stretching
20

21
Amber Force Field: Angle Bending
Amber force field treats angles that are bonded to the same central atom as springs (Hooke's Law, F = -kx) where Kθ is the empirical bending force constant, θ is the actual bond angle and θeq is the “natural” (empirical) bond angle 21
 where Kθ is the empirical bending force constant, θ is the actual bond angle and θeq is the natural (empirical) bond angle. 21.")
22
Amber Force Field: Angle Bending
22

23
Amber Force Field: Torsion Energy
Torsion Energy: torsional (dihedral) angle rotation between atoms that are vicinal (bonded to adjacent atoms) to each other where Vn is the barrier to free rotation for the “natural” bond, n is the periodicity of the rotation (number of cycles in 360°), φ is the torsion angle and γ is the angle where the potential passes through its minimum value 23
 angle rotation between atoms that are vicinal (bonded to adjacent atoms) to each other. where Vn is the barrier to free rotation for the natural bond, n is the periodicity of the rotation (number of cycles in 360°), φ is the torsion angle and γ is the angle where the potential passes through its minimum value. 23.")
24
Amber Force Field: Torsion Energy
24

25
Amber Force Field: Nonbonded Terms Enonbonded = Eelectrostatic + EvdW
The nonbonded Energy terms in Amber force field describe the long-range electrostatic and van der Waals interactions: Enonbonded = Eelectrostatic + EvdW 25

26
Amber Force Field: Electrostatic Energy
The Electrostatic Energy in the Amber force field represents the pair-wise sum of the electrostatic energies of all possible interacting non-bonded atoms i and j: where qi and qj are the point charges on atoms, Rij is the interatomic distance and ε is the dielectric constant 26

27
Amber Force Field: van der Waals Energy
The van der Waals Energy in the Amber force field represents the pair-wise sum of the van der Waals energies of all possible interacting non-bonded atoms i and j: where the Aij and Bij parameters control the depth and position (interatomic distance) of the potential energy well for a given pair of non-bonded interacting atoms and Rij is the interatomic distance 27
 of the potential energy well for a given pair of non-bonded interacting atoms and Rij is the interatomic distance. 27.")
28
Amber Force Field - Empirical Parameters

29
Where Do Empirical Parameters Come From?
29

30
Parameter Derivation: Partial Charges
Connolly In AMBER: Partial atomic charges are static Quantum chemical methods (B3LYP/ccpVTZ//HF/6-31G**) are used to generate an electrostatic potential (ESP) around a molecule on the spheric grid 3) RESP (Restrained Electrostatic Potential) Method is used to derive the partial charges QM = ab initio, DFT, semi-empirical
 are used to generate an electrostatic potential (ESP) around a molecule on the spheric grid. 3) RESP (Restrained Electrostatic Potential) Method is used to derive the partial charges. QM = ab initio, DFT, semi-empirical.")
31
Parameter Derivation: Van der Waals Parameters
It is the most difficult part… 1) Optimizing van der Waals parameters to reproduce the experimental or high-level Quantum Chemical data Could be computationally expensive 2) Optimizing van der Waals parameters through the Monte Carlo or MD simulations to reproduce the experimental properties of bulk solvent (density, etc.). For example, OPLS van der Waals parameters Could be computationally expensive 3) Reusing existing van der Waals parameters for similar atom types from the same or other force field The simplest approach
 Optimizing van der Waals parameters to reproduce the experimental or high-level Quantum Chemical data. Could be computationally expensive. 2) Optimizing van der Waals parameters through the Monte Carlo or MD simulations to reproduce the experimental properties of bulk solvent (density, etc.). For example, OPLS van der Waals parameters. Could be computationally expensive. 3) Reusing existing van der Waals parameters for similar atom types from the same or other force field. The simplest approach.")
32
Parameter Derivation: Bond and Angle Interactions
req and θeq come either from experimental data (X-ray, neutron diffraction) or Quantum Chemical calculations (geometry optimization) Kr and Kθ force constants are usually optimized to reproduce the vibration frequencies calculated using high-level Quantum Chemical methods. Or (the simplest approach) Kr and Kθ force constants could be derived from the existing bond/angle parameters for similar bond/angle types from the same or other force field
 or Quantum Chemical calculations (geometry optimization) Kr and Kθ force constants are usually optimized to reproduce the vibration frequencies calculated using high-level Quantum Chemical methods. Or (the simplest approach) Kr and Kθ force constants could be derived from the existing bond/angle parameters for similar bond/angle types from the same or other force field.")
33
Parameter Derivation: Dihedral Angle Interactions
Vn, n, and γ are derived to reproduce the rotational profile from the high-level Quantum Chemical calculations. Or (the simplest approach) Vn, n, and γ could be derived from the existing dihedral angle parameters for similar dihedral angle types from the same or other force field J.Wang et al., Development and testing of a general amber force field, Journal of Computational Chemistry, 25 (2004), 1157
 Vn, n, and γ could be derived from the existing dihedral angle parameters for similar dihedral angle types from the same or other force field. J.Wang et al., Development and testing of a general amber force field, Journal of Computational Chemistry, 25 (2004),")
34
Force Fields in Amber 12 ff99SB ff10 ff12SB Proteins ff99 +
backbone torsion Modifications no change from ff99SB + new backbone and sidechain torsions DNA ff99 “Barcelona” modifications RNA backbone changes + “OL3” changes for c J.Wang et al., Development and testing of a general amber force field, Journal of Computational Chemistry, 25 (2004), 1157
,")
35
Force Fields in Amber 12 Lipid11: A modular lipid force field
A new modular force field for the simulation of phospholipids and cholesterol designed to be compatible with the other pairwise additive Amber force field J.Wang et al., Development and testing of a general amber force field, Journal of Computational Chemistry, 25 (2004), 1157
,")
36
Other Force Fields in Amber: Inclusion of Polarization
Non-additive" force fields based on atom-centered dipole polarizabilities can also be used. These add a "polarization" term to what was given above where μi is an induced atomic dipole. In addition, charges that are not centered on atoms, but are off-center (as for lone-pairs or "extra points") can be included in the force field. 36
 can be included in the force field. 36.")
37
Other Force Fields in Amber: AMOEBA
(Atomic Multipole Optimized Energetics for Biomolecular Applications) Atomic Multipoles: The model uses a polarizable atomic multipole description of electrostatic interactions. Multipoles through the quadrupole are assigned to each atomic center based on a distributed multipole analysis (DMA) derived from large basis set molecular orbital calculations at the MP2/aug-cc-pVTZ level and the experimental geometry of the gas-phase monomer. Polarization is treated via self-consistent induced atomic dipoles. Atomic dipole polarizabilities can be derived from an empirical fit to experimentally known molecular polarizabilities. The induced dipole at each atomic site is computed as where αi is the atomic polarizability and Ei,α is the sum of the fields generated by both permanent multipoles and induced dipoles 37
 Atomic Multipoles: The model uses a polarizable atomic multipole description of electrostatic interactions. Multipoles through the quadrupole are assigned to each atomic center based on a distributed multipole analysis (DMA) derived from large basis set molecular orbital calculations at the MP2/aug-cc-pVTZ level and the experimental geometry of the gas-phase monomer. Polarization is treated via self-consistent induced atomic dipoles. Atomic dipole polarizabilities can be derived from an empirical fit to experimentally known molecular polarizabilities. The induced dipole at each atomic site is computed as. where αi is the atomic polarizability and Ei,α is the sum of the fields generated by both permanent multipoles and induced dipoles. 37.")
38
Other Force Fields: AMOEBA
(Atomic Multipole Optimized Energetics for Biomolecular Applications) The functional forms for bond stretching and angle bending were taken from the MM3 force field: A Urey-Bradley functional form was chosen for the stretch-bend term: 38
 The functional forms for bond stretching and angle bending were taken from the MM3 force field: A Urey-Bradley functional form was chosen for the stretch-bend term: 38.")
39
Other Force Fields: AMOEBA
(Atomic Multipole Optimized Energetics for Biomolecular Applications) Repulsion-Dispersion. The buffered 14-7 potential has been applied to model pairwise additive vdW interactions where εij is the potential well depth, ρij= Rij/R0ij with Rij as the i-j separation and R0ij the minimum energy distance. n = 14, m = 7, δ = 0.07, γ=0.12. The combining rules are: The buffered 14-7 function yields a repulsive region softer than the Lennard-Jones 6-12 function but steeper than typical Buckingham exp-6 formulations. The buffered 14-7 form was found to outperform Lennard-Jones and Buckingham potentials in simultaneously reproducing gas phase ab initio results and liquid thermodynamic properties of noble gases and a series of diatomic species. 39
 Repulsion-Dispersion. The buffered 14-7 potential has been applied to model pairwise additive vdW interactions. where εij is the potential well depth, ρij= Rij/R0ij with Rij as the i-j separation and R0ij the minimum energy distance. n = 14, m = 7, δ = 0.07, γ=0.12. The combining rules are: The buffered 14-7 function yields a repulsive region softer than the Lennard-Jones 6-12 function but steeper than typical Buckingham exp-6 formulations. The buffered 14-7 form was found to outperform Lennard-Jones and Buckingham potentials in simultaneously reproducing gas phase ab initio results and liquid thermodynamic properties of noble gases and a series of diatomic species. 39.")
40
What we can do with Amber?
40

41
Molecular Dynamics Simulations
The Molecular Dynamics simulation method is based on Newton’s second law or the equation of motion, F=ma, where F is the force exerted on the particle, m is its mass and a is its acceleration Integration of the equations of motion then yields a trajectory that describes the positions, velocities and accelerations of the particles as they vary with time. From this trajectory, the average values of properties can be determined. 41

42
MD: Melting of Ice 42

43
Human carboxyl esterasecomplexed with morphine
43

44
MD: Translocation of DNA
This movie shows the electrophoretically-driven translocation of a 58-nucleotid DNA strand through the transmembrane pore of alpha-hemolysin 44

45
Molecular Dynamics: Amber MD Workhorses
SANDER - Simulated Annealing with NMR-Derived Energy Restraints PMEMD - Particle Mesh Ewald Molecular Dynamics PMEMD is up to 55% faster than SANDER SANDER GPU PMEMD 45

46
Molecular Dynamics: What Are Current Simulation Capabilities?
Time scales of biological processes Femtosecond (fs) = second Picosecond (ps) = second Nanosecond (ns) = 10-9 second Microsecond (μs) = 10-6 second 46
 = second. Picosecond (ps) = second. Nanosecond (ns) = 10-9 second. Microsecond (μs) = 10-6 second. 46.")
47
Molecular Dynamics Trajectory Snapshots of Representative Structures
Molecular Dynamics Simulation Time Snapshots Snapshots of Representative Structures Molecular dynamics trajectory is a file containing snapshots of the simulated system 47

48
For each Snapshot Amber Saves Structure and Energy Decomposition
Energy Decomposition for each snapshot is written in the form: NSTEP = TIME(PS) = TEMP(K) = PRESS = Etot = EKtot = EPtot = BOND = ANGLE = DIHED = 1-4 NB = EEL = VDWAALS = EELEC = EHBOND = RESTRAINT = EKCMT = VIRIAL = VOLUME = Density = Ewald error estimate: E-03 Snapshots of Representative Structures
 = TEMP(K) = PRESS = Etot = EKtot = EPtot = BOND = ANGLE = DIHED = NB = EEL = VDWAALS = EELEC = EHBOND = RESTRAINT = EKCMT = VIRIAL = VOLUME = Density = Ewald error estimate: E Snapshots of Representative Structures.")
49
Plotting Molecular Dynamics Properties
Equilibration step allows atoms and molecules to find more natural positions with respect to one another Equilibration step MD Phase During the MD Phase molecular properties (structures, energies, etc.) are accumulated for future analysis
 are accumulated for future analysis.")
50
Not all system properties reach equilibrium at the same time

51
Trajectory snapshots after MD
Advanced MD Analysis in Amber Conformational clustering tools is available in ptraj ptraj uses several different algorithms for clustering trajectory frames into groups based on pairwise similarity Clustering Trajectory snapshots after MD 51

52
Statistical Ensembles
The microscopic state of a system is defined by the atomic positions, q, and momenta, p; these can also be considered as coordinates in a multidimensional space called phase space A single point in phase space, denoted by G, describes the state of the system An ensemble is a collection of points in phase space satisfying the conditions of a particular thermodynamic state. A Molecular Dynamics simulations generates a sequence of points in phase space as a function of time; These points belong to the same ensemble, and they correspond to the different conformations of the system and their respective momenta 52

53
Statistical Ensembles Supported in
Amber Microcanonical ensemble (NVE) : The thermodynamic state characterized by a fixed number of atoms, N, a fixed volume, V, and a fixed energy, E. This corresponds to an isolated system. Canonical Ensemble (NVT): This is a collection of all systems whose thermodynamic state is characterized by a fixed number of atoms, N, a fixed volume, V, and a fixed temperature, T. Isobaric-Isothermal Ensemble (NPT): This ensemble is characterized by a fixed number of atoms, N, a fixed pressure, P, and a fixed temperature, T. 53
 : The thermodynamic state characterized by a fixed number of atoms, N, a fixed volume, V, and a fixed energy, E. This corresponds to an isolated system. Canonical Ensemble (NVT): This is a collection of all systems whose thermodynamic state is characterized by a fixed number of atoms, N, a fixed volume, V, and a fixed temperature, T. Isobaric-Isothermal Ensemble (NPT): This ensemble is characterized by a fixed number of atoms, N, a fixed pressure, P, and a fixed temperature, T. 53.")
54
Advanced MD Techniques in Amber Adaptively Biased Molecular Dynamics (ABMD) method
ABMD is a method for the computation of the free energy surface of a reaction coordinate using non-equilibrium dynamics. Chemical reactions, conformational transitions, etc, occur when the system migrates from one local equilibrium minimum to another, overcoming the usually large energy barriers that separate reagents from products. The probability of such an event occurring spontaneously depends exponentially on the energy barrier and easily exceeds the computational time regime that present-day computer technology can afford. 54

55
Advanced Molecular Dynamics Techniques in Amber: Path integral molecular dynamics
Path integral molecular dynamics simulations can be used to sample equilibrium canonical distributions using quantum dynamics rather than Newton's equations for nuclear motion. Both equilibrium and kinetic isotope effects can be estimated via thermodynamic integration over mass. Centroid Molecular Dynamics (CMD) is an approximate method for calculating real- time quantum correlation functions. Ring Polymer Molecular Dynamics (RPMD). Both CMD and RPMD simulations provide an efficient route for the calculation of approximate correlation functions, which can then be related to the true quantum correlation functions. 55
 is an approximate method for calculating real- time quantum correlation functions. Ring Polymer Molecular Dynamics (RPMD). Both CMD and RPMD simulations provide an efficient route for the calculation of approximate correlation functions, which can then be related to the true quantum correlation functions. 55.")
56
How to Treat Bulk System?
Bulk (“infinite”) solvent We run computer simulation to predict and study the properties of a system in bulk (very big or “infinite” system) 56
 solvent. We run computer simulation to predict and study the properties of a system in bulk (very big or infinite system) 56.")
57
How to Treat Bulk System?
Treatable system But… We can simulate only a relatively small number of particles in order not to slow down the computation. Artificial surface effect Problem: we are not interested in surface effects 57

58
Possible Solutions? But… Such system is too big to simulate…
1) The system size should be extremely large to ensure that the surface has only a small influence on the bulk properties But… Such system is too big to simulate… Surface effect has small influence on the bulk properties 58
 The system size should be extremely large to ensure that the surface has only a small influence on the bulk properties. But… Such system is too big to simulate… Surface effect has small influence on the bulk properties. 58.")
59
Possible Solutions? 2) Surface effects can be ignored for all system sizes if we use periodic boundary conditions. The cubical simulation box (Central Box) is replicated throughout space to form an infinite lattice All other boxes are identical to the Central Box (its copies) Central Box No surface 59
 is replicated throughout space to form an infinite lattice. All other boxes are identical to the Central Box (its copies) Central Box. No surface. 59.")
60
Periodic Boundary Conditions
2) Then one of its images will enter through the opposite face 1) If a molecule leaves the Central Box 60
 Then one of its images will enter through the opposite face. 1) If a molecule leaves the Central Box. 60.")
61
Periodic Boundary Conditions
61

62
Periodic Boundary Conditions in AMBER
Rectangular parallelepiped 1) Truncated octahedron 2) Truncated octahedron has the advantage of being more nearly spherical than most other MD cells. This can be very useful when simulating a large molecule in solution, where fewer solvent molecules are required for a given simulation cell width. 62
 Truncated octahedron. 2) Truncated octahedron has the advantage of being more nearly spherical than most other MD cells. This can be very useful when simulating a large molecule in solution, where fewer solvent molecules are required for a given simulation cell width. 62.")
63
Estimation of Binding Energies in Non-Covalent complexes
In general, non-covalent bonding refers to attractive intermolecular forces that are not covalent in nature. Non-covalent interactions may include ionic bonds, hydrophobic interactions, hydrogen bonds and van der Waals forces. 63

64
Estimation of Binding Energies in Non-Covalent complexes
Protein-ligand complex 64

65
Evaluating Free Energies of Binding using Amber
65

66
Evaluating Free Energies of Binding: MM-PBSA
The acronym MM-PBSA stands for Molecular Mechanics- Poisson Bolzmann Surface Area The MM-PBSA approach represents the postprocessing method to evaluate free energies of binding or to calculate absolute free energies of molecules in solution. Acc. Chem. Res. 2000, 33, 66

67
Evaluating Free Energies of Binding: MM-PBSA
One carries out a molecular dynamics simulation, typically in a periodic box with water and counterions (“regular” MD simulation), and correct representation of long-range electrostatic effects such as PME, saving a set of representative structures. After MD Simulation any solvent and counterion molecules are removed, and the free energy, G, is calculated according to the following equation: 67
, and correct representation of long-range electrostatic effects such as PME, saving a set of representative structures. After MD Simulation any solvent and counterion molecules are removed, and the free energy, G, is calculated according to the following equation: 67.")
68
Evaluating Free Energies of Binding: MM-PBSA
any solvent and counterion molecules are removed 68

69
Evaluating Free Energies of Binding: MM_PBSA
where G is the calculated average free energy, and EMM is the average molecular mechanical energy: where these correspond to the bond, angle, torsion, van der Waals, and electrostatic terms in the molecular mechanical force field, evaluated with no nonbonded cutoff. 69

70
Evaluating Free Energies of Binding: MM_PBSA
GPBSA is the solvation free energy calculated with a numerical solution of the Poisson-Bolzmann equation and an estimate of the nonpolar free energy with a simple surface area term. -TSMM is the solute entropy, which can be estimated by quasi harmonic analysis of the trajectory or, in selected cases, by using normal-mode analysis. This final term is likely to be much smaller than the other two in many applications of estimating relative free energies. 70

71
Evaluating Free Energies of Binding: Thermodynamic integration
The thermodynamic integration (TI) technique allows to calculate the free energy difference between two systems, A and B, by slowly interconverting the Hamiltonian HA (representing system A) into the Hamiltonian HB (representing system B), during the course of the simulation. This process could involve the annihilation or creation of atoms (“Computational alchemy” ). Examples: Atom → nothing Group of Atoms (or Molecule) → nothing Charge on Atom → No charge on Atom Charge on Group of Atoms (or Molecule) → → No charge on Group of Atoms (Molecule) 71
 technique allows to calculate the free energy difference between two systems, A and B, by slowly interconverting the Hamiltonian HA (representing system A) into the Hamiltonian HB (representing system B), during the course of the simulation. This process could involve the annihilation or creation of atoms ( Computational alchemy ). Examples: Atom → nothing. Group of Atoms (or Molecule) → nothing. Charge on Atom → No charge on Atom. Charge on Group of Atoms (or Molecule) → → No charge on Group of Atoms (Molecule) 71.")
72
“Computational alchemy”
One common application of this model is pKa calculations, where the charges are mutated from the protonated to the deprotonated form Disappears during simulation 72

73
Evaluating Free Energies of Binding: Thermodynamic integration
The free energy difference is then given by The subscript λ at the pointed angles indicates that the average should be taken over an ensemble with Hamiltonian Hλ . In MD simulations the integral is often replaced by a sum over a discrete set of values of λ: where Δλ is chosen such that the result is statistically accurate while using a minimum of computer time. 73

74
Inclusion of Solvation Effects in Amber
Practically all important biological processes take place in solvent Solvation methods can be devided into two main categories: explicit (supermolecule) and implicit solvation methods 74
 and implicit solvation methods. 74.")
75
Explicit solvent model
Molecular solvent models employ hundreds or thousands of discrete solvent molecules Pros: Many of the properties of solutions and solutes can be reproduced Cons: Such calculations converge only slowly to precise answers because of the large number of particles and states involved; expensive computationally. 75

76
Implicit Solvation Methods in Amber
Implicit solvation schemes speed up the calculations by orders of magnitude and are assumed to compromise little on essential features of the solvation phenomenon. 76

77
Continuum solvation models
Continuum model treat the solvent as a continuous medium having the average properties of the real solvent and surrounding the solute beginning at or near its van der Waals or Solvent-accessible surface. Pros: Faster than molecular solvation models Cons: Obtaining accurate numerical solutions for a large system such as a protein still has a significant computational cost 77

78
Implicit Solvation Methods in Amber: The Generalized Born/Surface Area Model
To estimate the total solvation free energy of a molecule, ΔGsolv , one typically assumes that it can be decomposed into the "electrostatic" and "non-electrostatic“ parts: where ΔGnonel is the free energy of solvating a molecule from which all charges have been removed (i.e. partial charges of every atom are set to zero), and ΔGel is the free energy of first removing all charges in the vacuum, and then adding them back in the presence of a continuum solvent environment. ΔGnonel comes from the combined effect of two types of interaction: the favorable van der Waals attraction between the solute and solvent molecules, and the unfavorable cost of breaking the structure of the solvent (water) around the solute. 78
, and ΔGel is the free energy of first removing all charges in the vacuum, and then adding them back in the presence of a continuum solvent environment. ΔGnonel comes from the combined effect of two types of interaction: the favorable van der Waals attraction between the solute and solvent molecules, and the unfavorable cost of breaking the structure of the solvent (water) around the solute. 78.")
79
Implicit Solvation Methods in Amber: The Generalized Born/Surface Area Model
Calculating ΔGnonel: In the Amber code ΔGnonel is taken to be proportional to the total solvent accessible surface area (SASA) of the molecule, with a proportionality constant derived from experimental solvation energies of small non-polar molecules, and uses a fast Linear Combinations of Pairwise Overlaps (LCPO) algorithm [J. Comput. Chem. 20, (1999)] to compute an analytical approximation to the surface accessible area of the molecule. 79
 of the molecule, with a proportionality constant derived from experimental solvation energies of small non-polar molecules, and uses a fast Linear Combinations of Pairwise Overlaps (LCPO) algorithm [J. Comput. Chem. 20, (1999)] to compute an analytical approximation to the surface accessible area of the molecule. 79.")
80
Implicit Solvation Methods in Amber: The Generalized Born/Surface Area Model
Calculating ΔGel: Within Amber GB models, each atom in a molecule is represented as a sphere of radius ρi with a charge qi at its center; the interior of the atom is assumed to be filled uniformly with a material of dielectric constant of 1. The molecule is surrounded by a solvent of a high dielectric εw (80 for water at 300 K) where rij is the distance between atoms i and j, the Ri are the so-called effective Born radii of atoms i and j, and fgb is a certain smooth function of its arguments. A common choice of fgb is 80
 where rij is the distance between atoms i and j, the Ri are the so-called effective Born radii of atoms i and j, and fgb is a certain smooth function of its arguments. A common choice of fgb is. 80.")
81
Implicit Solvation Methods in Amber: ALPB (Analytical Linearized Poisson-Boltzmann)
Based on an approximate analytical solution of the linearized Poisson-Bolzmann equation for a sphere (Kirkwood, 1934). The basic ALPB equation that approximates the electrostatic part of the solvation free energy is: where β = εin /εex is the ratio of the internal and external dielectrics, α = , and A is the so-called effective electrostatic size of the molecule. fgb is the same smooth function as in the GB model. The GB approximation is then just the special case of ALPB when the solvent dielectric is infinite; however, for finite values of solvent dielectric the ALPB tends to be more accurate. Grigori Sigalov, Andrew Fenley, and Alexey Onufriev, J. Chem. Phys. 124, (2006) Grigori Sigalov, Peter Scheffel, and Alexey Onufriev, J. Chem. Phys. 122, (2005) 81
. The basic ALPB equation that approximates the electrostatic part of the solvation free energy is: where β = εin /εex is the ratio of the internal and external dielectrics, α = , and A is the so-called effective electrostatic size of the molecule. fgb is the same smooth function as in the GB model. The GB approximation is then just the special case of ALPB when the solvent dielectric is infinite; however, for finite values of solvent dielectric the ALPB tends to be more accurate. Grigori Sigalov, Andrew Fenley, and Alexey Onufriev, J. Chem. Phys. 124, (2006) Grigori Sigalov, Peter Scheffel, and Alexey Onufriev, J. Chem. Phys. 122, (2005) 81.")
82
Implicit Solvation Methods in Amber: Poisson-Boltzmann solver
An efficient finite-difference numerical solver is implemented for various applications of the Poisson-Boltzmann (PB) method. The electrostatic potential φj at atomic charge site is computed by solving the PB equation: where ε(r) is the dielectric constant, φ(r) is the electrostatic potential, ρ(r) is the solute charge, zi is the charge of ion type i, ci is the number density of ion type i far from the solute, kB is the Boltzmann constant, and T is temperature; the summation is over all different ion types. This is the most rigorous method for treatment of implicit solvent in Amber It can be used for both static (single point) and dynamic applications. However, it is much slower than GB and ALPB and memory intensive for macromolecules. 82
 method. The electrostatic potential φj at atomic charge site is computed by solving the PB equation: where ε(r) is the dielectric constant, φ(r) is the electrostatic potential, ρ(r) is the solute charge, zi is the charge of ion type i, ci is the number density of ion type i far from the solute, kB is the Boltzmann constant, and T is temperature; the summation is over all different ion types. This is the most rigorous method for treatment of implicit solvent in Amber. It can be used for both static (single point) and dynamic applications. However, it is much slower than GB and ALPB and memory intensive for macromolecules. 82.")
83
Inclusion of Solvation Effects in Amber: RISM
RISM - Reference Interaction Site Model RISM is an approximate solution to the Ornstein- Zernike (OZ) equation: where r12 is the separation between particles 1 and 2 while Ω1 and Ω2 are their orientations relative to the vector r12. The two functions in this relation are h, the total correlation function, and c, the direct correlation function. 83
 equation: where r12 is the separation between particles 1 and 2 while Ω1 and Ω2 are their orientations relative to the vector r12. The two functions in this relation are h, the total correlation function, and c, the direct correlation function. 83.")
84
RISM: Practical Considerations
Calculating a 3D-RISM solution for a single solute conformation typically requires about 100 times more computer time than the same calculation with explicit solvent or PB. Memory: anywhere from a few megabytes for the smallest solutes to gigabytes for large complexes 84

85
Exploring Conformational Space of Biomolecules
85

86
Conformational Space of Biomolecules Can Be Very Complex
86

87
Exploring Conformational Space of Biomolecules
Due to this property of the free energy landscape, efficient computational approaches for searching for low-energy minima in these complex systems present a great challenge. 87

88
Exploring Conformational Space: Simulating Annealing
Temperature Cooling phase Cooling phase Heating phase Heating phase Time Local Minima Energy Profile 88

89
Exploring Conformational Space: REMD
REMD stands for the Replica Exchange Method Dynamics In REMD several noninteracting copies (replicas) are independently and simultaneously simulated at different temperatures. Replica 1, T1 Replica 2, T2 Replica N, TN At intervals during the otherwise standard simulations, conformations of the system being sampled at different temperatures are exchanged based on a Metropolis-type criterion 89
 are independently and simultaneously simulated at different temperatures. Replica 1, T1. Replica 2, T2. Replica N, TN. At intervals during the otherwise standard simulations, conformations of the system being sampled at different temperatures are exchanged based on a Metropolis-type criterion. 89.")
90
Exploring Conformational Space: REMD
Replica 1, T1 Replica 2, T2 Replica N, TN As a result, the low temperature simulations (replicas) have the potential to escape kinetic traps by jumping to minima that are being sampled by the higher-temperature replicas where kinetic trapping is less prevalent. 90
 have the potential to escape kinetic traps by jumping to minima that are being sampled by the higher-temperature replicas where kinetic trapping is less prevalent. 90.")
91
Treating Long-Range Electrostatic Interactions
91

92
Treating Long-Range Electrostatic Interactions
Cut-off 92

93
Treating Long-Range Electrostatic Interactions
93

94
Treating Long-Range Electrostatic Interactions in Amber
The particle-mesh Ewald (PME) procedure (or, optionally, a "true" Ewald sum) is used to handle long-range electrostatic interactions. 94
 procedure (or, optionally, a true Ewald sum) is used to handle long-range electrostatic interactions. 94.")
95
Doing Semi-Empirical Quantum Chemistry with Amber
Amber 12 is packaged with sqm - a linear scaling semi-empirical program for calculation of energies, charges and geometries of systems up to ˜20,000 atoms. 95

96
Doing Semi-Empirical Quantum Chemistry with Amber
sqm’s Available features include: Linear scaling Divide and Conquer (D&C) calculations. Single point AM1, PM3, MNDO, MNDO/d or PDDG-PM3 calculations. Geometry Optimization (steepest decent, conjugate gradient, BFGS, and LBFGS available) Mulliken, CM1 and CM2 charge analysis Nuclear Magnetic Resonance prediction and simulation Mixed quantum mechanics/molecular mechanics (QM/MM) linear scaling Semi-Empirical calculations. 96
 calculations. Single point AM1, PM3, MNDO, MNDO/d or PDDG-PM3 calculations. Geometry Optimization (steepest decent, conjugate gradient, BFGS, and LBFGS available) Mulliken, CM1 and CM2 charge analysis. Nuclear Magnetic Resonance prediction and simulation. Mixed quantum mechanics/molecular mechanics (QM/MM) linear scaling Semi-Empirical calculations. 96.")
97
Doing Semi-Empirical Quantum Chemistry with Amber
Amber 12 is packaged with SQM semi- empirical program. MNDO: H, Li, Be, B, C, N, O, F, Al, Si, P, S, Cl, Zn, Ge, Br, Sn, I, Hg, Pb AM1: H, C, N, O, F, Al, Si, P, S, Cl, Zn, Ge, Br, I, Hg PM3: H, Be, C, N, O, F, Mg, Al, Si, P, S, Cl, Zn, Ga, Ge, As, Se, Br, Cd, In, Sn, Sb, PDDG/PM3: H, C, N, O, F, Si, P, S, Cl, Br, I PDDG/MNDO: H, C, N, O, F, Cl, Br, I RM1: H, C, N, O, P, S, F, Cl, Br, I PM3CARB1: H, C, O PM6: H, He, Li, Be, B, C, N, O, F, Ne, Na, Mg, Ar, K, Ca, Zn, Ga, Ge,Kr, Rb, DFTB/SCC-DFTB: (Any atom set available from the website) 97
 97.")
98
A Hybrid Quantum Mechanical/Molecular Mechanical (QM/MM) Approach
 Approach")
99
Why Do We Need a Hybrid QM/MM Approach?
Quantum Mechanics Molecular Mechanics generally applicable restricted to the classes of molecule it have been designed for allow the calculation of ground and excited state properties: molecular energies and structures, energies and structures of transition states, atomic charges, reaction pathways etc. allow the calculation of ground state properties: relative molecular energies and structures CPU and memory hungry. Computationally efficient Suitable for small and medium size systems Suitable for large molecular systems

100
Why Do We Need a Hybrid QM/MM Approach?
The main bottleneck of quantum chemical methods is that they are CPU and memory hungry. For example, for small peptide of 126 atoms one energy evaluation requires: CPU Time Memory Method Seconds Time units KB Memory units Quantum chemical* 273.00 1820 4889 85 Molecular Mechanical 0.15 1 58 *Semi-empirical PM3 method In general, CPU and memory requirements (N – number of atoms): Molecular Mechanical methods ~ N2 Semiempirical Quantum Chemical methods Ab initio Quantum Chemical methods ~ N4
: Molecular Mechanical methods. ~ N2. Semiempirical Quantum Chemical methods. Ab initio Quantum Chemical methods. ~ N4.")
101
A Hybrid QM/MM Approach
The general idea of a hybrid QM/MM approach is that large chemical systems may be partitioned into 1) an electronically important region (QM region) which requires a quantum chemical treatment and 2) a remainder which only acts in a perturbative fashion and thus admits a classical description (MM region).
 an electronically important region (QM region) which requires a quantum chemical treatment and 2) a remainder which only acts in a perturbative fashion and thus admits a classical description (MM region).")
102
The Simplest Hybrid QM/MM Model
Hamiltonian for molecular system in the Born-Oppenheimer approximation: “Standard” QM hamiltonian The MM region is viewed in the QM calculations as a set of point charges The main drawbacks of this simple QM/MM model are: it is impossible to optimize the position of the QM part relative to the external charges because QM nuclei will collapse on the negatively charged external charges. some MM atoms possess no charge and so would be invisible to the QM atoms the van der Waals terms on the MM atoms often provide the only difference in the interactions of one atom type versus another, i.e. chloride and bromide ions both have unit negative charge and only differ in their van der Waals terms.

103
A Hybrid QM/MM Model So, it is quite reasonable to attribute the van der Waals parameters (as it is in the MM method) to every QM atom and the Hamiltonian describing the interaction between the QM and MM atoms can have a form: The van der Waals term models also electronic repulsion and dispersion interactions, which do not exist between QM and MM atoms because MM atoms possess no explicit electrons. A. Warshel, M. Levitt // Theoretical Studies of Enzymic Reactions: Dielectric, Electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. // J.Mol.Biol. 103(1976),
 to every QM atom and the Hamiltonian describing the interaction between the QM and MM atoms can have a form: The van der Waals term models also electronic repulsion and dispersion interactions, which do not exist between QM and MM atoms because MM atoms possess no explicit electrons. A. Warshel, M. Levitt // Theoretical Studies of Enzymic Reactions: Dielectric, Electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. // J.Mol.Biol. 103(1976),")
104
The Hybrid QM/MM Model Now we can construct a “real” hybrid QM/MM Hamiltonian: A “standard” MM force field can be used to determine the MM energy. For example, AMBER-like force field has a form:

105
Choice of QM method ... is a compromise between computational efficiency and practicality and the desired chemical accuracy. The main advantage of semi-empirical QM methods is that their computational efficiency is orders of magnitude greater than either the density functional or ab initio methods Ab initio method Semi-empirical method

106
Calibration of the QM/MM potential
Crucial aspect is how the interaction between QM and MM parts is determined. In choosing the appropriate form, it is required that the balance between attractive and repulsive forces must be preserved and the QM/MM interactions must be of the correct magnitude with respect to the separate QM and MM contributions

107
Calibration of the QM/MM potential: Parameterizations
1) 2) 1) Modification of the one-electron terms arising from interaction of the electron cloud of the QM fragment with the point charge of an MM atom. 2) By varying the radii in the van der Waals terms. 3) By varying 1)+2)
 2) 1) Modification of the one-electron terms arising from interaction of the electron cloud of the QM fragment with the point charge of an MM atom. 2) By varying the radii in the van der Waals terms. 3) By varying 1)+2)")
108
Calibration of the QM/MM potential
1) By hand, to find the optimum values of the parameters by calculating interaction curves for charge/ion systems and comparing them with the MP2/ G** ab initio results. M.J. Field, P.A. Bash, M. Karplus, J.Comp.Chem., 11(1990), 2) Fitting calculated H-bond energies to experimental data on ion-molecular complexes in the gas phase. V.V. Vasilyev, A.A. Bliznyuk, A.A. Voityuk, Int.J.Quant.Chem. 44(1992),
 By hand, to find the optimum values of the parameters by calculating interaction curves for charge/ion systems and comparing them with the MP2/ G** ab initio results. M.J. Field, P.A. Bash, M. Karplus, J.Comp.Chem., 11(1990), ) Fitting calculated H-bond energies to experimental data on ion-molecular complexes in the gas phase. V.V. Vasilyev, A.A. Bliznyuk, A.A. Voityuk, Int.J.Quant.Chem. 44(1992),")
109
Calibration of the QM/MM potential
3) Optimizing van der Waals parameters on QM atoms to reproduce the 6- 31G(d) interaction energies for H-bonded complexes in the gas phase. P.A. Bash, L. Lawrence, A.D. MacKerell, Jr., D. Levine, P. Hallstrom, PNAS USA, 93(1996), 4) Optimizing van der Waals parameters on QM atoms to reproduce the MP2/6-31G(dp) interaction energies for H-bonded complexes in the gas phase. J. Gao // Toward a molecular Orbital Derived Empirical Potential for Liquid Simulations // J.Phys.Chem. B 101(1997), 5) By varying the radii in the van der Waals terms to reproduce experimental free energies of solvation using MD simulations. P.L. Cummins, J.E. Gready, J.Comp.Chem., 18(1997),
 Optimizing van der Waals parameters on QM atoms to reproduce the 6- 31G(d) interaction energies for H-bonded complexes in the gas phase. P.A. Bash, L. Lawrence, A.D. MacKerell, Jr., D. Levine, P. Hallstrom, PNAS USA, 93(1996), ) Optimizing van der Waals parameters on QM atoms to reproduce the MP2/6-31G(dp) interaction energies for H-bonded complexes in the gas phase. J. Gao // Toward a molecular Orbital Derived Empirical Potential for Liquid Simulations // J.Phys.Chem. B 101(1997), ) By varying the radii in the van der Waals terms to reproduce experimental free energies of solvation using MD simulations. P.L. Cummins, J.E. Gready, J.Comp.Chem., 18(1997),")
110
Dividing Covalent Bonds across the QM and MM Regions
In many simulations it is necessary to have the QM/MM boundary cut covalent bonds, and a number of additional approximations have to be made.

111
Dividing Covalent Bonds across the QM and MM Regions
Using a hybrid orbital on the frontier MM atom A. Warshel, M. Levitt // Theoretical Studies of Enzymic Reactions: Dielectric, Electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. // J.Mol.Biol. 103 (1976), V. Thery, D. Rinaldi, J.-L. Rivail, B. Maigret, G.G. Ferenczy, J.Comp.Chem. 15 (1995), 269
, V. Thery, D. Rinaldi, J.-L. Rivail, B. Maigret, G.G. Ferenczy, J.Comp.Chem. 15 (1995), 269.")
112
Dividing Covalent Bonds across the QM and MM Regions
Using “link” atoms “Link” atoms are used to gracefully cap the electron density. This approach is used in Amber

113
Implementation of “link” Atom Approach in Amber 9 & 10
The link atom is placed along the bond vector joining the QM and MM atom The default link atom type is hydrogen It interacts with MM region only electrostatically (no VDW term). WdV interaction between QM and MM atoms which form 1-2 and 1-3 “bonded” pairs is not calculated. Bond stretching, angle bending, and torsion interactions between QM and MM regions are calculated as those in MM if 1-2, 1-2-3, or terms contain at least one MM atom
. WdV interaction between QM and MM atoms which form 1-2 and 1-3 bonded pairs is not calculated. Bond stretching, angle bending, and torsion interactions between QM and MM regions are calculated as those in MM if 1-2, 1-2-3, or terms contain at least one MM atom.")
114
Reviews on QM/MM H. Hu and W. Yang, Free energies of chemical reactions in solution and in enzymes with ab initio quantum mechanics/molecular mechanics methods, Annu Rev Phys Chem. 2008;59: C. Bo and F. Maseras, QM/MM methods in inorganic chemistry, Dalton Trans., 2008, 2911–2919 H.M. Senn and W. Thiel, QM/MM studies of enzymes, Current Opinion in Chemical Biology, 2007(11), R.A. Friesner and V. Guallar, Ab initio Quantum Chemical and Mixed Quantum Mechanics/Molecular Mechanics (QM/MM) Methods for Studying Enzymatic Catalysis, Annual Review of Physical Chemistry, 2005 (56), G. Monard, X. Prat-Resina, A. González-Lafont, J.M. Lluch, Determination of enzymatic reaction pathways using QM/MM methods, Int. J Quant Chem, 2003, 93 Issue 3, Pages
, R.A. Friesner and V. Guallar, Ab initio Quantum Chemical and Mixed Quantum Mechanics/Molecular Mechanics (QM/MM) Methods for Studying Enzymatic Catalysis, Annual Review of Physical Chemistry, 2005 (56), G. Monard, X. Prat-Resina, A. González-Lafont, J.M. Lluch, Determination of enzymatic reaction pathways using QM/MM methods, Int. J Quant Chem, 2003, 93 Issue 3, Pages")
115
Hints for running QM/MM calculations Choosing the QM region
There are no good universal rules here One might want to have as large a QM region as possible However, having more than atoms in the QM region will lead to simulations that are very expensive.

116
Hints for running QM/MM calculations Choosing the QM region
For many features of conformational analysis, a good MM force field may be better than a semi- empirical or DFTB quantum description.

117
Hints for running QM/MM calculations Choosing the QM region

118
QM Methods in Amber 12 Available semi- empirical Hamiltonians are MNDO, AM1, PM3, RM1, PDDG/PM3, PDDG/MNDO, and PM3CARB1, PM3- MAIS, MNDO/d, AM1/d (Mg from AM1/d and H, O, and P from AM1/d-PhoT) and PM6 They can be used for gas phase, generalized Born and PME periodic simulations.
 and PM6. They can be used for gas phase, generalized Born and PME periodic simulations.")
119
QM Methods in Amber 12 Support is also available the DFT methods:
The Density Functional Theory-based-tight- binding (DFTB) Hamiltonian The Self-Consistent-Charge version, SCC-DFTB In Amber 9 the DFTB/SCC-DFTB implementation does not support generalized Born, PME or Ewald calculations,
 Hamiltonian. The Self-Consistent-Charge version, SCC-DFTB. In Amber 9 the DFTB/SCC-DFTB implementation does not support generalized Born, PME or Ewald calculations,")
120
The elements supported by QM methods in Amber 12
MNDO: H, Li, Be, B, C, N, O, F, Al, Si, P, S, Cl, Zn, Ge, Br, Sn, I, Hg, Pb AM1: H, C, N, O, F, Al, Si, P, S, Cl, Zn, Ge, Br, I, Hg PM3: H, Be, C, N, O, F, Mg, Al, Si, P, S, Cl, Zn, Ga, Ge, As, Se, Br, Cd, In, Sn, Sb, Te, I, Hg, Tl, Pb, Bi PDDG/PM3: H, C, N, O, F, Si, P, S, Cl, Br, I PDDG/MNDO: H, C, N, O, F, Cl, Br, I PM3CARB1: H, C, O DFTB/SCC-DFTB: H, C, N, O, S, Zn

121
QM/MM calculations: ab initio and DFT methods
Amber can support QM/MM simulations via an interface to external QM software packages: ADF (Amsterdam Density Functional) Gaussian GAMESS-US Orca NWChem TeraChem
 Gaussian. GAMESS-US. Orca. NWChem. TeraChem.")
122
QM/MM calculations: ab initio and DFT methods
Mechanical and electrostatic embedding: Gaussian Orca TeraChem Mechanical embedding: ADF GAMESS-US NWChem

123
Importance of Visualization
One quick look at the structure can help to detect errors and save days or weeks of your time

124
Freeware Visualization Programs: RasMol

125
Freeware Visualization Programs: VMD (Visual Molecular Dynamics)
VMD is a molecular visualization program for displaying, animating, and analyzing large biomolecular systems using 3-D graphics and built-in scripting.

126
Freeware Visualization Programs: gOpenMol

128
Freeware Visualization Programs: Chimera

129
Freeware Visualization Programs: MD Display
A Multi-platform 3D Stereo Molecular Dynamics Trajectory Visualization Package

130
Commercial Programs … they represent an expert molecular modeling environment which provides construction, editing, and visualization tools for both large and small molecules Tripos ( Accelrys ( and others…

131
Learning Amber

132
Amber Basic Tutorials http://ambermd.org/tutorials/
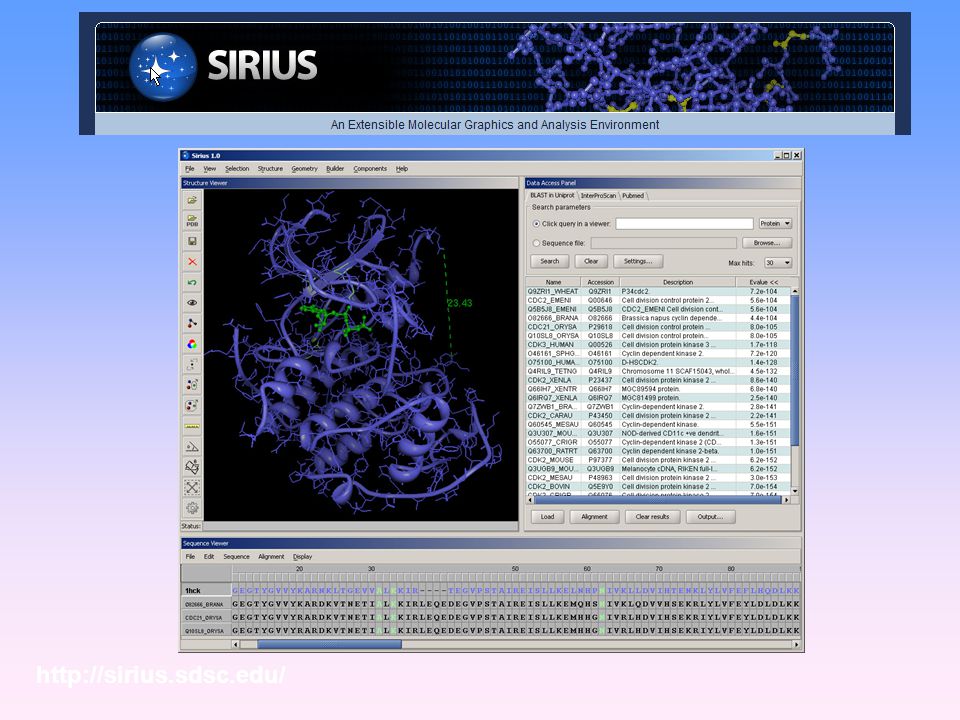
Simulating a small fragment of DNA Basic introduction to LEaP, sander, and ptraj, to build, solvate, run MD and analyze trajectories. Using VMD with AMBER Brief introduction to using VMD for visualising AMBER inpcrd, restrt and trajectory files Folding TRP Cage Vreating structures using XLeap followed by running heating and long MD simulations to conduct protein folding experiments. Advanced analysis: RMSd fitting, mdcrd to binpos conversion, average structure calculation, hydrogen bond analysis and dihedral angle tracking using ptraj Demo of Ptraj Commands How to use AMBER's ptraj analysis program to analyse a peptide simulation and gather a range of statistics from the trajectory. Visualizing Amber Trajectories with Sirius how to use Sirius visualization software to display and analyze AMBER MD trajectory files 132

133
Amber Advanced Tutorials
Setting up an Advanced System (Including Charge Derivation) Preparing a system, for simulation with sander, that contains several non-standard residues A simple coupled potential QM/MM/MD simulation. How to set up a simple QM/MM/MD simulation of NMA in solution using AMBER 9 MM-PBSA Step by step explanation of using the mm_pbsa script in AMBER 9 to calculate the binding energy of the RAS-RAF protein complex Nudged Elastic Band (NEB) method How use the NEB method to predict a pathway for a conformational change in alanine dipeptide. pKa Calculations using Thermodynamic Integration How to calculate the pKa value of the ASP residue in the protein thioredoxin … and other 133
 Preparing a system, for simulation with sander, that contains several non-standard residues. A simple coupled potential QM/MM/MD simulation. How to set up a simple QM/MM/MD simulation of NMA in solution using AMBER 9. MM-PBSA. Step by step explanation of using the mm_pbsa script in AMBER 9 to calculate the binding energy of the RAS-RAF protein complex. Nudged Elastic Band (NEB) method. How use the NEB method to predict a pathway for a conformational change in alanine dipeptide. pKa Calculations using Thermodynamic Integration. How to calculate the pKa value of the ASP residue in the protein thioredoxin. … and other")
134
Resume Amber package represents an expert molecular modelling environment with a reach functionality and good computer performance. 134

135
Hands-on Web: sf.anu.edu.au/~vvv900/monash
Tutorial files (AMBER_INTRO_COURSE/): standard-setup.tar – “Standard” setup (long) nonstandard.tar – Handling non-standard residues (long) amber-gaussian.tar – QM/MM using Amber- Gaussian interface (short) qm-mm.tar – QM/MM using Amber inbuilt semiempirical methods 135
: standard-setup.tar – Standard setup (long) nonstandard.tar – Handling non-standard residues (long) amber-gaussian.tar – QM/MM using Amber- Gaussian interface (short) qm-mm.tar – QM/MM using Amber inbuilt semiempirical methods")
Similar presentations


 Workshop.>")

 potential QM/MM/MD simulations in Amber Dr. Vladislav Vasilyev Supercomputer Facility The Australian National University.>")





: Reading: Van Holde, Chapter 1 Van Holde Chapter 3.1 to 3.3 Van Holde Chapter 2 (we’ll go through Chapters 1 and 3 first. 1.Van.>")





 EXAMPLE I: THE PROTEIN FOLDING PROBLEM Alexey Onufriev, Virginia.>")


