Download presentation
Presentation is loading. Please wait.

1
Susan Burner Bankowski, M.S., J.D. Chair, OHSU IRB
New IRB Policies Reporting Unanticipated Problems & Adverse Events Data & Safety Monitoring Susan Burner Bankowski, M.S., J.D. Chair, OHSU IRB

2
Old Policy Everything and the Kitchen Sink. Lacks Context and Meaning
Results in waste of resources & arbitrary revisions to consent form

3
PI Reporting Requirements
To the IRB All Unanticipated Problems involving risks to human subjects or others. 45 CFR (b)(5). To the Sponsor Any adverse effect that may reasonably be regarded as caused by, or probably caused by, the drug. If the adverse effect is "alarming," the investigator must report the adverse effect immediately (§ (b))
(5). To the Sponsor. Any adverse effect that may reasonably be regarded as caused by, or probably caused by, the drug. If the adverse effect is alarming, the investigator must report the adverse effect immediately (§ (b))")
4
IRB Reporting Requirements
An assured institution must have - Written procedures for ensuring prompt reporting to the IRB, appropriate institutional officials, and the department or agency head of (i) any unanticipated problems involving risks to subjects or others… CFR (b)(5). OHSU IRB Reports to IO and OHRP.
 any unanticipated problems involving risks to subjects or others….. 45 CFR (b)(5). OHSU IRB Reports to IO and OHRP.")
5
Sponsor’s Reporting Requirements
To the PI “Keep each participating investigator informed of new observations discovered by or reported to the sponsor on the drug, particularly with respect to adverse effects and safe use” (§ (b)) Notify investigators of any adverse experience “associated with the drug that is both serious and unexpected” and “any finding from tests in laboratory animals that suggest a significant risk for human subjects,” 21 C.F.R. § (c)(1)(i)(A), (B); To the FDA For devices - any “unanticipated adverse device effect” 21 C.F.R. § (b), 21 C.F.R. § (b)(1). Safety and effectiveness data (annual reports & IND safety reports)
) Notify investigators of any adverse experience associated with the drug that is both serious and unexpected and any finding from tests in laboratory animals that suggest a significant risk for human subjects, 21 C.F.R. § (c)(1)(i)(A), (B); To the FDA. For devices - any unanticipated adverse device effect 21 C.F.R. § (b), 21 C.F.R. § (b)(1). Safety and effectiveness data (annual reports & IND safety reports)")
6
What is an Adverse Event?
Any untoward or undesirable, although not necessarily unexpected, event experienced by a human subject that may be a result of: The interventions and interactions use in the research The collection of identifiable private information in the research An underlying disease, disorder, or condition of the subject ; and/or Other circumstances unrelated to the research or any underlying disease, disorder, or condition of the subject.

7
What is a Serious Adverse Event?
Any AE that is: Is fatal Is life-threatening Is persistent or significantly disabling or incapacitating Results in inpatient hospitalization or prolongation of hospitalization Results in psychological or emotional harm requiring treatment Creates a persistent or significant disability Causes a congenital anomaly or birth defect and/or Results in a significant medical incident

8
What is an Unanticipated Problem?
Events that are not expected given the nature of the research procedures and the subject population being studied and suggest that the research places subjects or others at a greater risk of harm or discomfort related to the research than was previously known or recognized. Harm to a subject need not occur for an event to be an unanticipated problem.

9
All AEs are not UPs! What to Report?
Do not Report A to IRB – only B &C Unanticipated Problems A = AEs that are not UPs B = AEs that are UPs C = UPs that are not AEs

10
When is an AE an UP? In order for the PI (or the Monitor) to determine whether a particular AE is also considered a UP, the following should be taken into account: The description of known or foreseeable adverse events and risks in the IRB-approved research protocol, any applicable investigator brochure, the current IRB-approved consent form, and other relevant sources of information. Any underlying disease or conditions of the subject experiencing the adverse event. A careful assessment of whether the adverse event is related or possibly related to the subject’s participation in the study.
 to determine whether a particular AE is also considered a UP, the following should be taken into account: The description of known or foreseeable adverse events and risks in the IRB-approved research protocol, any applicable investigator brochure, the current IRB-approved consent form, and other relevant sources of information. Any underlying disease or conditions of the subject experiencing the adverse event. A careful assessment of whether the adverse event is related or possibly related to the subject’s participation in the study.")
11
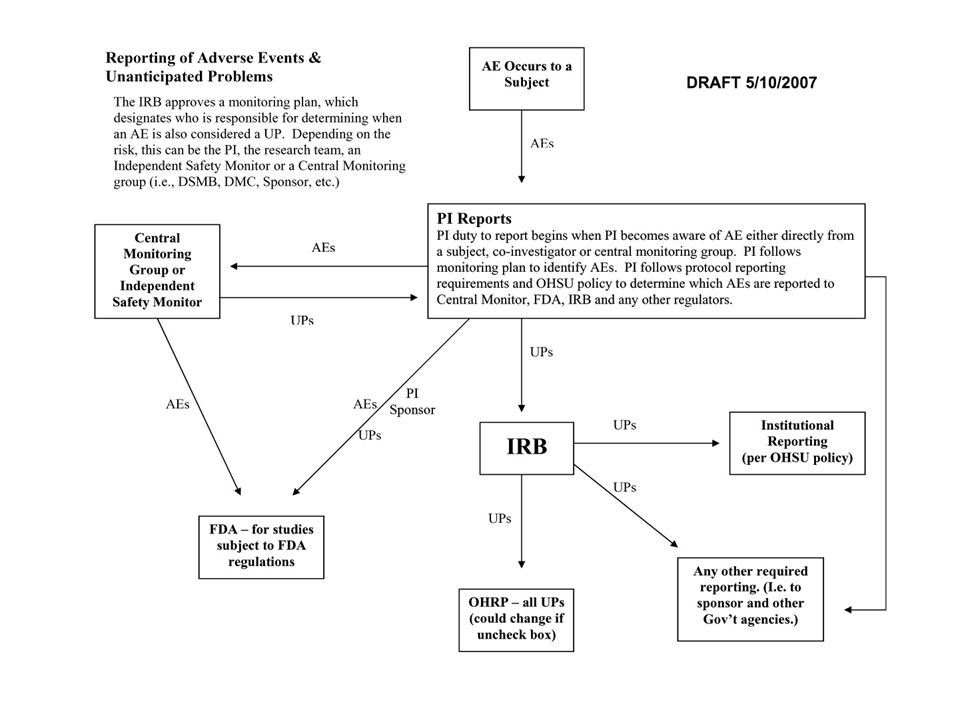
Unanticipated Problem Analysis Chart

12
Categories of Reportable AEs
On Protocol SAEs that are unanticipated and related or possibly related to the research participation. On Protocol SAEs or AEs that are anticipated and related or possibly related, but are occurring at a significantly higher frequency or severity than expected. On or Off Protocol Unanticipated SAEs or AEs that are related or possibly related, regardless of severity, that may alter the risks for subjects and, as a result, warrant changes to the protocol and/or consent process On or Off Protocol other events that are unanticipated that may place subjects or others at a greater risk of harm or discomfort than was previously known or recognized. Harm to a subject need not have occurred.

13
What’s Meant by Unexpected?
Any AE may be considered unexpected if it occurs in one or more subjects, the nature, severity, or frequency of which is not consistent with either: the known or foreseeable risk of AEs associated with the procedures involved in the research that are described in the protocol-related documents, such as the IRB-approved research protocol, any applicable investigator brochure, and the current IRB-approved informed consent document, and other relevant sources of information, such as product labeling and package inserts; or the expected natural progression of any underlying disease, disorder, or condition of the subject(s) experiencing the adverse event and the subject’s predisposing risk factor profile for the adverse event.
 experiencing the adverse event and the subject’s predisposing risk factor profile for the adverse event.")
14
What’s Meant by Possibly Related?
Adverse events may be caused by one or more of the following: the procedures involved in the research; an underlying disease, disorder, or condition of the subject; or other circumstances unrelated to either the research or any underlying disease, disorder, or condition of the subject. However, AEs that are determined to be at least partially caused by (1) would be considered related to participation in the research, whereas adverse events determined to be solely caused by (2) or (3) would be considered unrelated to participation in the research.
 would be considered related to participation. in the research, whereas adverse events determined to be. solely caused by (2) or (3) would be considered unrelated. to participation in the research.")
15
Who is doing the Monitoring?
Not the IRB! A Monitoring Entity Investigator Monitor Independent Monitor Data Safety Monitoring Board (DSMB)/Data Monitoring Committee (DMC) In a multi-site study, there must be a monitoring entity that is central to all sites.
/Data Monitoring Committee (DMC) In a multi-site study, there must be a monitoring entity that is central to all sites.")
16
All should have a Centralized Monitor.
It is the monitor’s responsibility to analyze and review applicable Non-OHSU AEs and Different Protocol Events, determine if the AEs are UPs, and report the UPs to the PI for subsequent reporting to the PIs IRB. If OHSU is serving as a coordinating center, then the PI of the coordinating center is responsible for this determination via an approved monitoring plan.

17
Monitoring Provisions
All research requires some level of monitoring and principal investigators are responsible for monitoring their studies. However, the IRB must approve the plan for monitoring data and safety for all research except minimal risk research where OHSU is the only site The monitoring provisions should be tailored to the expected risks of the research, the type of subject population being studied and the nature, size and complexity of the research protocol. The monitoring provisions must be described in sufficient detail, including the type of monitoring entity, for the IRB to determine whether they are appropriate for the research.

18
Monitoring Entities Investigator Monitor – The Principal Investigator or a Co-investigator who is responsible for data and safety monitoring. Independent Monitor - A qualified and objective individual or group not directly involved with the design and conduct of the study (e.g., safety officer, designated Medical Monitor or Monitoring Group). These individuals may or may not be employees of OHSU or the study sponsor. However, conflict of interest is an important consideration when employees of the study sponsor have the primary responsibility for monitoring data from the standpoint of scientific integrity and participant safety.
. These individuals may or may not be employees of OHSU or the study sponsor. However, conflict of interest is an important consideration when employees of the study sponsor have the primary responsibility for monitoring data from the standpoint of scientific integrity and participant safety.")
19
Monitoring Entities Continued
Data Safety Monitoring Board (DSMB)/Data Monitoring Committee (DMC) – an independent formal committee that is established specifically to monitor data throughout the life of a study to determine if it is appropriate, from both the scientific and ethical standpoint, to continue the study as planned. DSMBs/DMCs are typically made up of individuals who have expertise in the field, experience in the conduct of clinical trials, and/or statistical knowledge, and who do not have any serious conflicts of interest, such as financial interests that could be substantially affected by the outcome of the trial, strong views on the relative merits of the interventions under study, or relationship with the sponsor or those in trial leadership positions that could be considered reasonably likely to affect their objectivity.
/Data Monitoring Committee (DMC) – an independent formal committee that is established specifically to monitor data throughout the life of a study to determine if it is appropriate, from both the scientific and ethical standpoint, to continue the study as planned. DSMBs/DMCs are typically made up of individuals who have expertise in the field, experience in the conduct of clinical trials, and/or statistical knowledge, and who do not have any serious conflicts of interest, such as financial interests that could be substantially affected by the outcome of the trial, strong views on the relative merits of the interventions under study, or relationship with the sponsor or those in trial leadership positions that could be considered reasonably likely to affect their objectivity.")
Similar presentations
 is a review committee established to help protect the rights and welfare of human research subjects.>")


















