Download presentation
Presentation is loading. Please wait.

1
Microarrays & Gene Expression Analysis

3
Contents DNA microarray technique Why measure gene expression
Clustering algorithms Relation to Cancer SAGE SBH – Sequencing By Hybridization

4
DNA Microarrays Developed around 1987.
Employ methods previously exploited in immunoassay context – specific binding and marking techniques. Two types of probes: Format I: probe cDNA (500~5,000 bases long) is immobilized to a solid surface such as glass; widely considered as developed at Stanford University; Traditionally called DNA microarrays. Format II: an array of oligonucleotide (20~80-mer oligos) probes is synthesized either in situ (on-chip) or by conventional synthesis followed by on-chip immobilization; developed at Affymetrix, Inc. Many companies are manufacturing oligonucleotide based chips using alternative in-situ synthesis or depositioning technologies. Historically called DNA chips.
 is immobilized to a solid surface such as glass; widely considered as developed at Stanford University; Traditionally called DNA microarrays. Format II: an array of oligonucleotide (20~80-mer oligos) probes is synthesized either in situ (on-chip) or by conventional synthesis followed by on-chip immobilization; developed at Affymetrix, Inc. Many companies are manufacturing oligonucleotide based chips using alternative in-situ synthesis or depositioning technologies. Historically called DNA chips.")
5
DNA Microarray Technique
The microarray is made of a small piece of glass (1x1 or 2x2 cm). Thousands to millions of pixels are put on it, in each many (n) copies of DNA probes (short (8-30 bases), single stranded, called OLIGO). A probe on the array will bind its complementary target if it is present in the solution washing the chip. When the array surface is scanned with a laser, fluorescent labels attached to the targets reveal which probes are bound.
. Thousands to millions of pixels are put on it, in each many (n) copies of DNA probes (short (8-30 bases), single stranded, called OLIGO). A probe on the array will bind its complementary target if it is present in the solution washing the chip. When the array surface is scanned with a laser, fluorescent labels attached to the targets reveal which probes are bound.")
6
Use of DNA Microarrays Identify a query sequence - the sequence is hybridized to an array containing suitable probes Point mutations (SNP) or other mutations – the array contains probes that match segments of the normal and mutated sequences. An unknown sequence (SBH) – the array contains all possible k-mers (e.g., all the 46 6-mers) Gene expression analysis - which genes are expressed ? under what conditions ?
 or other mutations – the array contains probes that match segments of the normal and mutated sequences. An unknown sequence (SBH) – the array contains all possible k-mers (e.g., all the 46 6-mers) Gene expression analysis - which genes are expressed under what conditions")
7
DNA Microarray Methodology - Flash Animation

9
Why Measure Gene Expression

10
Why Measure Gene Expression
Determines which genes are induced/repressed in response to a developmental phase or to an environmental change.

11
Why Measure Gene Expression
Determines which genes are induced/repressed in response to a developmental phase or to an environmental change. Sets of genes whose expression rises and falls under the same condition are likely to have a related function.

12
Why Measure Gene Expression
Determines which genes are induced/repressed in response to a developmental phase or to an environmental change. Sets of genes whose expression rises and falls under the same condition are likely to have a related function. Features such as a common regulatory motif can be detected within co-expressed genes.

13
Why Measure Gene Expression
Determines which genes are induced/repressed in response to a developmental phase or to an environmental change. Sets of genes whose expression rises and falls under the same condition are likely to have a related function. Features such as a common regulatory motif can be detected within co-expressed genes. A pattern of gene expression may be used as an indicator of abnormal cellular regulation. A useful tool for cancer diagnosis

14
Clustering Co-expressed Genes
Find genes whose expression rises and falls under the same conditions. Methods include: Hierarchical clustering. Self organizing maps. Support vector machines (SVMs).
.")
15
Hierarchical Clustering
Cluster analysis and display of genome-wide expression patterns. Michael B. Eisen, Paul T. Spellman, Patrick O. Brown , and David Botstein, 1998, Relationships among objects (genes) are represented by a tree whose branch lengths reflect the degree of similarity between the objects, as assessed by a pairwise similarity function. The computed trees can be used to order genes in the original data table, so that genes or groups of genes with similar expression patterns are adjacent.
 are represented by a tree whose branch lengths reflect the degree of similarity between the objects, as assessed by a pairwise similarity function. The computed trees can be used to order genes in the original data table, so that genes or groups of genes with similar expression patterns are adjacent.")
16
GeneExplorer GeneCards pointer UniGene pointer Zoom:

17
Similarity Metric The gene similarity metric is a form of correlation coefficient. Let Gi equal the (log-transformed) primary data for gene G in condition i. For any two genes x and y observed over a series of N conditions, a similarity score can be computed as follows: S(x,y) = i=1..N(xi-x)(yi-y) / (std(x)*(std(y)) where x,y are the mean of observations on genes x and y. A neighbor joining method is used to built the corresponding tree.
 primary data for gene G in condition i. For any two genes x and y observed over a series of N conditions, a similarity score can be computed as follows: S(x,y) = i=1..N(xi-x)(yi-y) / (std(x)*(std(y)) where x,y are the mean of observations on genes x and y. A neighbor joining method is used to built the corresponding tree.")
18
Tree Creation For any set of n genes, a similarity matrix is computed by using the metric described above. The matrix is scanned to identify the highest value (representing the most similar pair of genes). A node is created joining these two genes, and a gene expression profile is computed for the node by averaging observation for the joined elements (missing values are omitted and the two joined elements are weighted by the number of genes they contain). The similarity matrix is updated with this new node replacing the two joined elements, and the process is repeated n-1 times until only a single element remains.
. A node is created joining these two genes, and a gene expression profile is computed for the node by averaging observation for the joined elements (missing values are omitted and the two joined elements are weighted by the number of genes they contain). The similarity matrix is updated with this new node replacing the two joined elements, and the process is repeated n-1 times until only a single element remains.")
19
Five separate clusters are indicated by colored bars and by identical coloring of the corresponding region of the dendrogram. The sequence-verified named genes in these clusters contain multiple genes involved in (A) cholesterol biosynthesis, (B) the cell cycle, (C) the immediate-early response, (D) signaling and angiogenesis, and (E) wound healing and tissue remodeling. These clusters also contain named genes not involved in these processes and numerous uncharacterized genes.
 cholesterol biosynthesis, (B) the cell cycle, (C) the immediate-early response, (D) signaling and angiogenesis, and (E) wound healing and tissue remodeling. These clusters also contain named genes not involved in these processes and numerous uncharacterized genes..")
20
Self Organizing Maps K-means method: the number of clusters is fixed (k). g1, ..,gn represents the expression of each gene gi in d experiments as a point in d dimensions. Randomly choose k centers, c1, ..,ck: ci is a point in a d dimension. The protocol: Join gi to the closest center. Compute new centers. The new center ci‘ is the center of mass of all points joined to ci. Repeat the steps until convergence or until you’re pleased with the results.

21
Relation to Cancer Tumors result from disruptions of growth regulation. Although most tumors are treated with general anti-proliferate drugs, they exhibit remarkable clinical heterogeneity which remains a major challenge in the successful management of cancer. Clinical heterogeneity in tumors likely reflects unrecognized molecular heterogeneity in tumors. Because of the logical connection between gene expression patterns and phenotype, it is likely that there is a direct connection between gene expression patterns of tumors and their clinical phenotype.

22
Towards a clinically relevant taxonomy of Cancer
Access archived clinical tumor samples taken at or near diagnosis from patients with well-characterized subsequent clinical histories. Use DNA arrays to measure gene expression in these samples. Look for new molecularly defined groups within or between previously recognized groups of tumors, especially groups with increased clinical homogeneity. Look for direct associations between molecular and clinical properties of tumors.

23
Cancer Gene Expression
The suggested procedure has been used to classify several types of cancer, or cancerous verses normal cells. Breast cancer AML and ALL. Melanoma. Lymphoma. …

24
Example - Melanoma Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 2000 Aug 3;406(6795):536-40 Discovered a subset of melanomas identified by mathematical analysis of gene expression in a series of samples.

25
Example - Melanoma Remarkably, many genes underlying the classification of this subset are differentially regulated in invasive melanomas that form primitive tubular networks in vitro, a feature of some highly aggressive metastatic melanomas. Global transcript analysis can identify unrecognized subtypes of cutaneous melanoma and predict experimentally verifiable phenotypic characteristics that may be of importance to disease progression.

26
Detection of Regulatory Motifs
A group of co-expressed genes is likely to be co-regulated during transcription. Transcription initiation is mediated by regulatory proteins that usually bind upstream to the transcription start site. The regulatory proteins bind to conserved regulatory motifs, a short DNA sequence. The upstream region of co-expressed genes can be searched for a common regulatory motif.

27
Other Applications – Predictive Tools
There is a correlation between co-expression and related gene function. “Inferring subnetworks from perturbed expression profiles.” Bioinformatics Jun;17 Suppl 1:S215-S224. There is a correlation between co-expression and protein-protein interaction. “Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae.” Nat Genet Dec;29(4):482-6. Poor correlation between gene expression and protein expression.
: Poor correlation between gene expression and protein expression.")
28
Correlation between gene and protein expression
Ideker et al., science 2001

29
Design & Probe Selection
Sensitivity – probes need to hybridize to their targets. For example – they need to avoid highly structured regions of the target molecule. Specificity – probes need not hybridize to wrong targets (cross hybridization). To this end: design probes to be long enough for statistical protection. search databases to explicitly avoid cross-hybridization to known foreign mRNA. Mismatch control.
. To this end: design probes to be long enough for statistical protection. search databases to explicitly avoid cross-hybridization to known foreign mRNA. Mismatch control.")
30
Other Challenges Analyze image to infer expression levels from red to green ratios, clean background, check for outliers, etc. Infer causal relations between genes – regulatory networks.

31
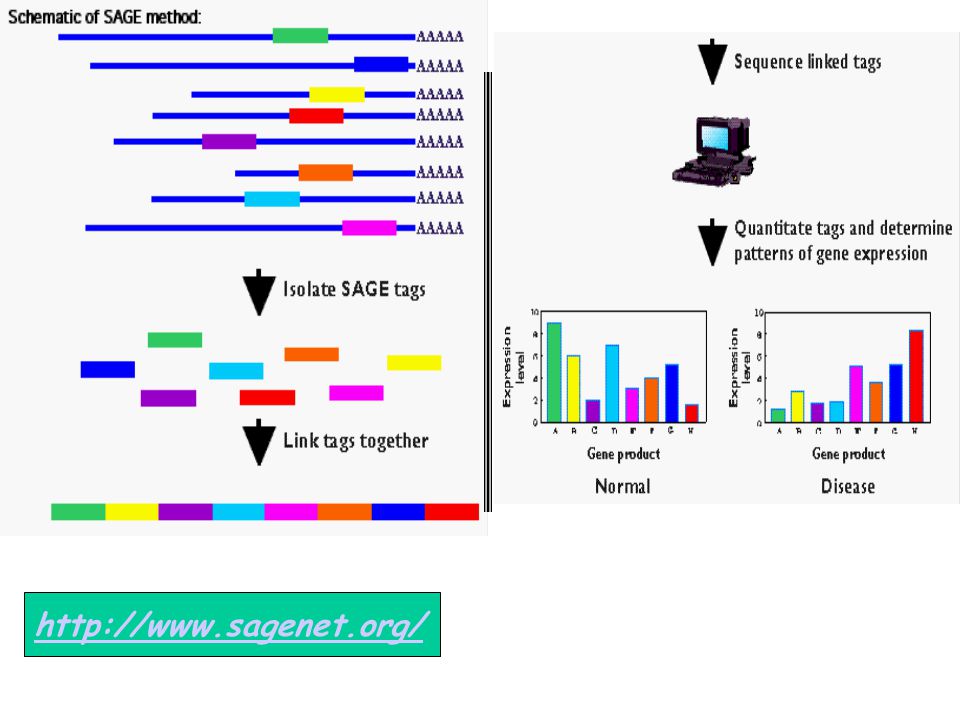
represents the transcription product of a gene.
Experimental technique assigned to gain a quantitive measure of gene expression. ~10-20 base “tags” are produced (immediately adjacent to the 3’ end of the 3’ most NlaIII restriction site). The SAGE technique measures not the expression level of a gene, but quantifies a "tag" which represents the transcription product of a gene.
. The SAGE technique measures not the expression level of a gene, but quantifies a tag which. represents the transcription product of a gene.")
32
SAGE Technique Extracting unique tagging sequences from mRNA molecules (tags are ~10-20b long). Concatenating the tags to a long sequence. Sequencing the resulting sequence and inferring levels from frequencies. Advantage: an unbiased and inclusive analysis of the transcriptome. Sequencing errors are especially problematic when tags are used, because of the short length of tags. Of roughly 1.5 million transcript sequences stored in GenBank, only about 180,000 are well characterized, and tags could represent them.

34
Colon cancer vs normal colon
A Colon cancer B Normal colon Colon cancer vs normal colon

36
SBH – Sequencing by Hybridization
A method for sequencing, actually the original motivation of DNA microarrays. A chip containing all k-mers is produced. The query sequence is hybridized to the chip. Example: a chip of all 3-mers is produced, containing 64 probes. 5 probes will be highlighted. C A T A T A Using chips for sequencing T A G A G T G T A C A T A G T A

37
SBH Protocol Knowing the start and end of the query sequence, and the set of highlighted k-mers, the query sequence is reconstructed. Example: start = CAT, end = GTA, highlighted group = {CAT, ATA, TAG, AGT, GTA}. CAT – AT? CAT ATA – TA? CATA TAG – AG? CATAG AGT – GTA CATAGT Problems: Reconstruction is not always unique – same k-mer may be followed by several k-mers. CAT – ATA, ATG. Hybridization contain errors.

Similar presentations















 A microarray may contain thousands of ‘spots’. Each spot contains many copies of the same DNA sequence that uniquely represents a gene from.>")


>")



