Download presentation
Presentation is loading. Please wait.

2
History/Background ‘Southern’ hybridization named after Sir Edwin Southern Developed in 1975 One of the most highly cited scientific publications Earned Sir Southern a Lasker Award in 2005

3
Southern Blotting Detection of specific sequences among DNA fragments separated by gel electrophoresis. o Restriction fragments appear as a smear. o DNA is transferred to the membrane the fragments retain the same pattern of separation o appear as bands.

4
Step 1. Restriction Enzyme Digestion EcoR I

5
Step 1. Restriction Enzyme Digestion

6
PROTOCOL AGAROSE GEL ELECTROPHORESIS Submerge the electrophorized gel in 0.2N HCl and agitate gently on a shaker, blue band turns green. Pour off the HCl and rinse the gel with deionized water. Cut off the top right corner of the agarose gel to simplify orientation during succeeding operations. Soak the gel in Denaturation Buffer for 30 minutes with gentle agitation. Rinse gel with dH 2 O soak in Neutralization Buffer- 30 minutes. Cut 3 pieces of (Whatman) filter paper to the size of the gel and 1 piece long enough to reach the bottom of the inverted gel tray.
 filter paper to the size of the gel and 1 piece long enough to reach the bottom of the inverted gel tray..")
7
Step 2. Gel Electrophoresis

8
Flow chart of Southern hybridization Preparing the samples and running the gel Southern transfer Probe preparation Prehybridization Hybridization Post-hybridization washing Signal detection Isotope Non-isotope

9
SOUTHERN TRANSFER Cut 1 piece of positively charged nylon membrane to the exact size of the gel. While the DNA is denaturing, Prepare; Invert gel tray in a large glass baking dish with the long piece of filter paper over it. The ends of the filter paper should drop to the bottom of the dish. Fill the dish with Transfer Buffer (20X SSC). Place 1 small piece of filter paper (wet with Transfer Buffer) on top of the gel tray. Remove the gel from the Neutralization Buffer and place it inverted on the filter paper.
. Place 1 small piece of filter paper (wet with Transfer Buffer) on top of the gel tray. Remove the gel from the Neutralization Buffer and place it inverted on the filter paper..")
10
Surround but do not cover the gel, Parafilm. Wet the top of the gel with Transfer Buffer. Soak nylon membrane in dH 2 O and place on top of the gel. To avoid bubbles, touch one corner of the membrane to the gel and gently lower the membrane onto the gel. Place the two filter papers on top of the membrane. Cut or fold a stack of paper towels (5-8 cm. high) and place them on top of the two filter papers. Place a glass plate and a beaker containing water on top of the stack to weigh the stack down. Allow the transfer of DNA to continue for 8-24 hours.
 and place them on top of the two filter papers. Place a glass plate and a beaker containing water on top of the stack to weigh the stack down. Allow the transfer of DNA to continue for 8-24 hours..")
12
Southern hybridization Transfer buffer

13
The next day, remove the weight, glass plate and paper towels. Lift the filter paper, the nylon membrane and the gel together, then invert. Mark the positions of the gel wells on the membrane with a soft lead pencil. Peel the membrane from the gel and rinse in 6X SSC for 10 minutes Wrap membrane in saran wrap and place it in the UV crosslinker for 2 minutes to crosslink DNA to membrane. Roll nylon membrane into a small roll and place inside a roller tube with the side containing the DNA facing the inside.

14
PRERHYBRIDISATION and RADIOLABELING OF PROBE : Preparing probe: Combine 10 µl template DNA & 6 µl dH2O. Denature the DNA by heating in a boiling water bath for 10 minutes and quickly chilling on ice. Mix DIG-High Prime thoroughly and add 4 µl to the denatured DNA, mix & centrifuge briefly. Incubate for 1 hour at 37ºC. Stop the reaction by adding 2 µl 0.5M EDTA. Add 15 ml DIG Easy Hybrid probe to the roller tube. Place the tube in the hybridizer for 30 minutes at 45ºC.

15
Step 6. Pre-hybridization Prehybridization buffers contain ‘blocking reagents’ that occupy available binding sites on the membrane

16
WASHING Discard probe. Add a preheated mixture of 4 ml DIG Easy Hybrid and 10 ml DNA to the tube. Put the tube back into the hybridization chamber at 65ºC overnight. The next day pour off the probe. Wash membrane twice for 5 min. each time in 2X Wash solution at room temperature. Wash membrane twice for 5 min. In prewarmed 0.1X or 0.5X Wash solution at 68ºC. Equilibrate membrane in Wash buffer for 2 min. at room temperature. Incubate membrane in 30 ml Maleic buffer for 30 min. at room temperature with gentle rotation.

17
DETECTION Remove membrane, add 3 µl Anti-Digoxigenin-AP, Fab Fragments, replace membrane and rotate gently for 30 minutes. Wash membrane twice for 15 min. Equilibrate membrane in Detection buffer for 2 min. Place membrane (nucleic acid side up) on parafilm. Spot 1 ml CSPD/Detection buffer solution over the membrane. Cover with another layer of parafilm and seal the edges. Incubate for 5 min. Tape membrane into pre-warmed x-ray cassette and incubate at 37ºC for 10 minutes.
 on parafilm. Spot 1 ml CSPD/Detection buffer solution over the membrane. Cover with another layer of parafilm and seal the edges. Incubate for 5 min. Tape membrane into pre-warmed x-ray cassette and incubate at 37ºC for 10 minutes..")
18
In the darkroom under a safelight, cut autorad x-ray film to appropriate size and tape over membrane Close cassette and expose for 15 minutes. Remove the film from the cassette under a safelight. Develop for 1 minute, fix for 3 minutes, then rinse twice with water and hang to dry.

19
No banding is seen although the gel showed presence of DNA. Probe may not have been good. Exposure times may have been incorrect (only 5 min. & overnight were used due to time constraints).
..")
20
Study gene expression by detection of RNA or (isolated mRNA) Observe cellular control over structure and function by determining particular gene expression. Capillary transfer of RNA from electrophoresis gel to the blotting membrane Developed by James Alwine, David Kemp and George Stark at University of Stanford

22
“It has not escaped our notice that the specific pairing we have postulated immediately suggests a copying mechanism for the genetic material.”

25
RNA is normally a single stranded molecule But, if we make an artificial complementary strand, it loves to be a double helix, just like its big sister, DNA. In this experiment we exploit this by making a specific complementary RNA strand to our gene. We label the complementary strand, this allows us to detect it and thus the mRNA with the target sequence

26
Classic procedure: Radioactive labelling of complementary RNA

27
U

28
The Probe: 1.Artificial RNA 2.Labelled with DIG 3.Produced in vitro

30
NBT + BCIP + alkaline phosphatase = color

32
RNA/DNA hybridization is influenced by… Probe concentration Sequence (GC bonds more stable than AT) Stringency
 Stringency")
33
Probe concentration: The higher the probe concentration, the faster the hybridization occurs. However, the higher the probe concentration, the higher the blot background (i.e. probe will stick everywhere). Low probe concentrations and long incubations (overnight) usually produce the best results.
. Low probe concentrations and long incubations (overnight) usually produce the best results..")
34
Stringency Stringency is a key concept in all nucleic acid detection experiments We can adjust the stringency so that only exact DNA or RNA sequences match. Or, we can allow some mismatching. We do this mostly by modifying wash conditions, especially salt. Salt: High salt wash, hybrids are more stable, more RNA stuck, blot is less stringent. Low salt wash, hybrids are less stable, blot is more stringent. Temperature: nucleic acids denature (helix melts) at high temperatures. High temperature, less stable, more stringent
 at high temperatures. High temperature, less stable, more stringent.")
35
We know the sequence of the target gene, X (more on this later in the course). Our probe is an exact match to it. So, we are using high stringency. Two washes, one high salt then one low salt. High temperature (65 C).
..")
36
SDS-PAGE and Western blotting

37
SDS-PAGE (PolyAcrylamide Gel Electrophoresis) The purpose of this method is to separate proteins according to their size, and no other physical feature.
 The purpose of this method is to separate proteins according to their size, and no other physical feature.")
38
SDS breaks up hydrophobic areas and coats proteins with negative charges thus overwhelming positive charges in the protein. The detergent binds to hydrophobic regions in a constant ratio of about 1.4 g of SDS per gram of protein.

40
The end result has two important features: 1) all proteins contain only primary structure 2) All proteins have a large negative charge which means they will all migrate towards the positive pole when placed in an electric field.
 all proteins contain only primary structure 2) All proteins have a large negative charge which means they will all migrate towards the positive pole when placed in an electric field.")
41
Before proteins are applied to the gel they are exposed to a disulfide bond reducing agent, mercaptoethanol. Mercaptoethanol denatures protein-reduces disulfide bonds

42
Polyacrylamide gels are prepared by free radical polymerization of acrylamide and the cross linking agent N,N’-methylene-bis- acrylamide. Chemical polymerization is controlled by an initiator catalyst system, N,N,N’,N’-tetramethylethylenediamine (TEMED)
.")
43
The copolymerization of acrylamide with methylenebisacrylamide produces a mesh- like network in three dimensions, consisting of acrylamide chains with interconnections formed from the methylenebisacrylamide.

45
The Electrophoresis Matrix In gel electrophoresis the matrix forces sample components to separate by size, as they move through its porous structure. The matrix provides greater resistance to the movement of larger molecules. the matrix serves as a solid medium upon which samples can be fixed and detected in post-electrophoretic analysis.

46
What happens after electrophoresis 1. Fix the proteins in the gel and them stain them. 2. Electrophorectic transfer to a membrane (Western blotting) and then probe with antibodies.
 and then probe with antibodies..")
47
Fixing (or fixation) -proteins are denatured and precipitated -insoluble aggregates -gel matrix. Fixation prevents the diffusion of proteins. Fixation removes gel buffer components, SDS, which may interfere in the staining process.

48
Fixing Proteins on Electrophoresis Gels Protein gels can be fixed effectively by soaking for 1 hr in 45% methanol, 45% water, and 10% glacial acetic acid. Solution is stable for up to 30 days at room temperature. A more stable fixative is 25% isopropanol, 65% water and 10% acetic acid, which can be stored at 4 o C for up to 4 months.

49
Staining Proteins in Gels Chemical stains detect proteins -differential binding of the stain by the protein molecules and the gel matrix. Nonspecific in action, detecting proteins without - individual identities. Characteristics of stain: low background, high sensitivity, large linear range and ease of use.

50
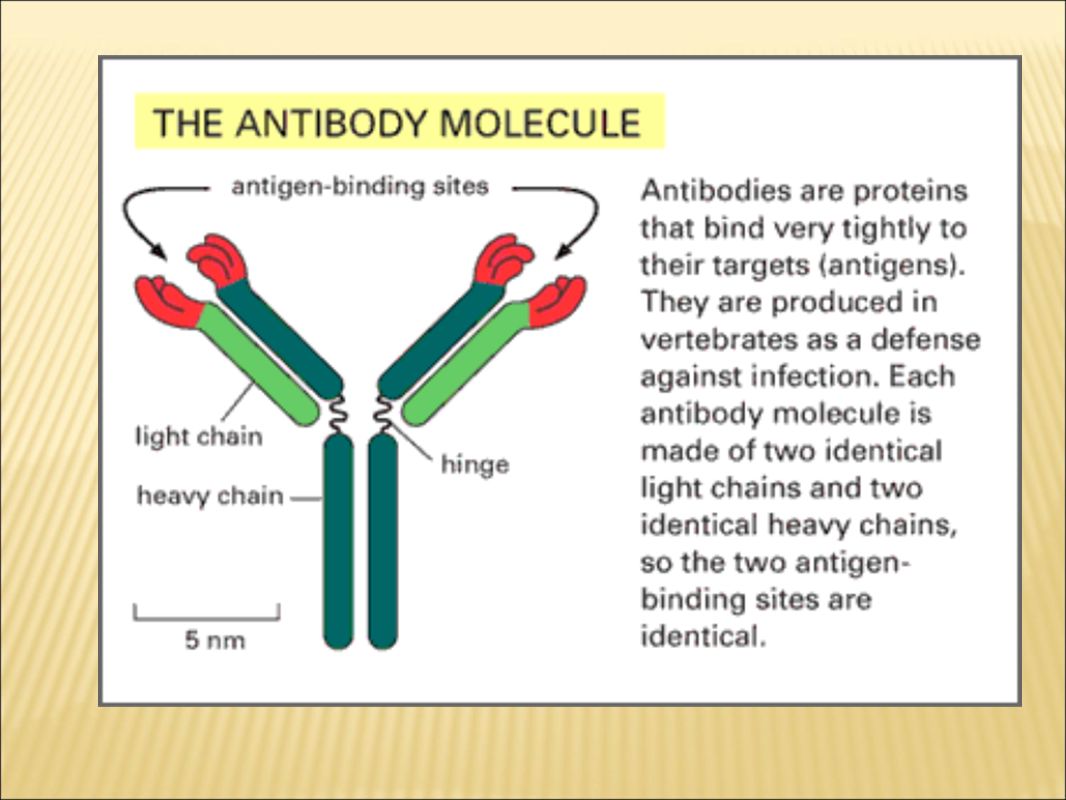
Western blotting Western blot analysis can detect one protein in a mixture of any number of proteins while giving you information about the size of the protein. Dependent on the use of a high-quality antibody directed against a desired protein. Use antibody as a probe to detect the protein of interest.

51
1. Place a nitrocellulose membrane on the gel. The actual blotting process may be active (electoblotting) or passive (capillary). Passive transfer - time consuming takes 1-2 days for complete protein transfer. Faster and efficient transfer by electroblotter. Sandwich of filter paper, gel, membrane and more filter paper is prepared in a cassette, which is placed between platinum electrodes. An electric current is passed through the gel causing the proteins to electrophorese out of the gel and onto the membrane.
 or passive (capillary). Passive transfer - time consuming takes 1-2 days for complete protein transfer. Faster and efficient transfer by electroblotter. Sandwich of filter paper, gel, membrane and more filter paper is prepared in a cassette, which is placed between platinum electrodes. An electric current is passed through the gel causing the proteins to electrophorese out of the gel and onto the membrane..")
52
3. Blocking - Ab = proteins, proteins bind to nitrocellulose. So before adding primary antibody, mask the nitrocellulose by incubating in a blot. Blotting agents around is Carnation Nonfat Dry Milk. 4. Incubate nitrocellulose membrane with primary Ab Ab specific for the protein of interest. The primary antibody binds to the desired protein forming an antigen- antibody complex. No visible signal. 5. Incubate nitrocellulose membrane,secondary antibody. Ab,antibody-enzyme conjugate. The secondary antibody should be an antibody against the primary antibody. Secondary antibody will "stick" to the primary antibody, just like the primary antibody "stuck" to the protein. The conjugated enzyme allows to visualize all of this..

54
6. Enzyme in action, incubate it in a reaction mix that is specific for your enzyme. Bands occur where there is a protein-primary antibody-secondary antibody-enzyme complex. Detection, the secondary antibody- tagged with enzyme - horseradish peroxidase (HRP) or alkaline phosphatase (AP). Blot is incubated in a substrate solution the conjugated enzyme catalyzes the conversion of the substrate into a visible product that precipitates. Coloured band indicates the position. Modification to the colored reactions using chemiluminescent substrates, converted by enzymes into products, generate a light signal, captured on film
 or alkaline phosphatase (AP). Blot is incubated in a substrate solution the conjugated enzyme catalyzes the conversion of the substrate into a visible product that precipitates. Coloured band indicates the position. Modification to the colored reactions using chemiluminescent substrates, converted by enzymes into products, generate a light signal, captured on film.")
56
Technique to analyse proteins, lipids or glycoconjugates and is most often used to detect carbohydrate epitope. Transferred proteins are analysed for PTM using probes that may detect lipids, carbohydrates, phosphorelation, or any other protein modification using probes.

57
Detection of protein modification in 2 bacterial species. Cholera toxin B subunit ( which bind to gangliosides ), Concanavalin A (which detects mannose-containing glycans ) and nitrophospho molybdate-methyl green ( which detects phosphoproteins) were used to detect proteins.
, Concanavalin A (which detects mannose-containing glycans ) and nitrophospho molybdate-methyl green ( which detects phosphoproteins) were used to detect proteins..")
58
Most proteins are translated from mRNA undergo modifications before becoming functional in cells. Expression of post translated proteins are important in several diseases.

59
To detect biomolecules. The culture containing molecule is applied in membrane as a dot. Detection – nucleotide probes or Ab Radioactive sample hybridised to detect variation between samples

Similar presentations



























![Lab#6 Western Blotting BCH 462[practical].](/18/6070629/big_thumb.jpg "Lab#6 Western Blotting BCH 462[practical].>")
 Northern blot (RNA) Western blot (Protein) Eastern blot (???)>")
>")
