Download presentation
Presentation is loading. Please wait.

1
ELIZABETH GARRETT-MAYER (SOME SLIDES BY PAT LORUSSO OF KARMANOS CANCER INSTITUTE WAYNE STATE UNIVERSITY) Phase I Trials of Chemotherapy and Targeted Agents
 Phase I Trials of Chemotherapy and Targeted Agents")
2
Phase I Trials PHASE I TRIALS ARE CLINICAL EXPERIMENTS If you don’t think your experiment through well before embarking on it, if you don’t identify the variables and account for them, the experiment may not be a success TEAM APPROACH You need a research team, including preclinical scientists, pharmacologists, medical oncologists, biostatisticians, to plan your study.

3
Phase I Trials MUST BE REALISTIC WITH QUESTIONS YOU WANT TO ANSWER MANY QUESTIONS IN PHASE I ARE CURRENTLY EXPLORATORY DUE TO SEVERAL FACTORS Limited patient numbers Multiple doses with small patient numbers Unknown assay Heterogeneous patient population

4
First evaluation of a new cancer therapy in humans ◦ First-in-human, first-in-kind (e.g. the first compound ever evaluated in humans against a new molecular target), single-agent ◦ First-in-human, but not first-in-kind (i.e. others agents of the same class have entered human testing), single-agent Definition(s) of a Phase I Trial
, single-agent ◦ First-in-human, but not first-in-kind (i.e. others agents of the same class have entered human testing), single-agent Definition(s) of a Phase I Trial.")
5
Multiple types of Phase I Trials ◦ Investigational agent + investigational agent ◦ Investigational agent + approved agent(s) ◦ Approved agent + approved agent(s) ◦ Approved or investigational agent with pharmacokinetic focus (adding of CYP inhibitor) ◦ Typically considered drug-drug interaction study ◦ Approved or investigational agent with pharmacodynamic focus (e.g. evaluation using functional imaging) ◦ Approved or investigational agent with radiotherapy ◦ Food effect study ◦ QTc prolongation study ◦ Bioequivalence study Definition(s) of a Phase I Trial
 ◦ Approved or investigational agent with radiotherapy ◦ Food effect study ◦ QTc prolongation study ◦ Bioequivalence study Definition(s) of a Phase I Trial.")
6
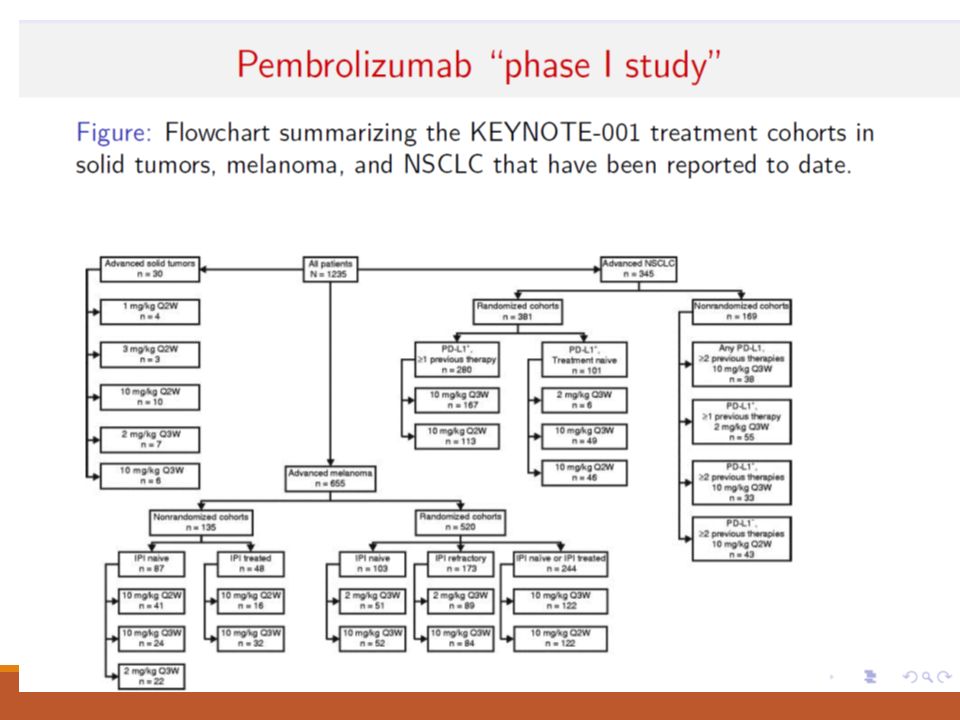
We are in a revolutionary state of affairs in phase I in cancer Nivolumab: Phase I trial ended up with ~300 patients Pembrolizumab: Phase I trial ended up with ~1300 patients. Novel immunotherapies are not ‘behaving’ like traditional anti-cancer therapies. Studies designs have changed but it’s not clear how to most efficiently tackle the first two phases of drug development with these novel agents.

7
Primary objective: ◦ Identify dose-limiting toxicities (DLTs) and the recommended phase II dose(s) (RP2D) Newer Primary objective: ◦ Identify the optimal dose—induces a clinical effect and operates with acceptable toxicity Secondary objectives: ◦ Define toxicity profile of new therapy(s) in the schedule under evaluation ◦ Pharmacokinetics (PK) ◦ Pharmacodynamic (PD) effects in tumor and/or surrogate tissues ◦ Antitumor activity Objectives of a Phase I Trial
 and the recommended phase II dose(s) (RP2D) Newer Primary objective: ◦ Identify the optimal dose—induces a clinical effect and operates with acceptable toxicity Secondary objectives: ◦ Define toxicity profile of new therapy(s) in the schedule under evaluation ◦ Pharmacokinetics (PK) ◦ Pharmacodynamic (PD) effects in tumor and/or surrogate tissues ◦ Antitumor activity Objectives of a Phase I Trial")
8
Classic Phase I Assumption: Efficacy and toxicity both increase with dose DLT = dose- limiting toxicity

9
Dose-limiting toxicity (DLT): ◦ Toxicity that is considered unacceptable (due to severity and/or irreversibility) and limits further dose escalation ◦ Defined with standard criteria CTCAE 4.1 ◦ Dose-limiting toxicity ◦ defined in advance prior to beginning the trial ◦ is protocol-specific ◦ Typically defined based on toxicity seen in the first cycle ◦ With select agents that have a more delayed toxicity (eg 2 nd /3 rd cycle), time allowed for DLT definition being re- evaluated Definitions of Key Concepts in Phase I Trials
: ◦ Toxicity that is considered unacceptable (due to severity and/or irreversibility) and limits further dose escalation ◦ Defined with standard criteria CTCAE 4.1 ◦ Dose-limiting toxicity ◦ defined in advance prior to beginning the trial ◦ is protocol-specific ◦ Typically defined based on toxicity seen in the first cycle ◦ With select agents that have a more delayed toxicity (eg 2 nd /3 rd cycle), time allowed for DLT definition being re- evaluated Definitions of Key Concepts in Phase I Trials")
10
Examples of DLTs – chronic (daily) dosing: ◦ Threshold for DLTs is lower ◦ Some Grade 2 toxicities may be unacceptable and intolerable due to their persistence and lack of time period for recovery ◦ Examples: ◦ Grade 2 intolerable or worse non-hematologic toxicity despite supportive measures ◦ Grade 3 or worse hematologic toxicity ◦ Inability to complete a pre-specified percentage of treatment during the cycle due to toxicity (e.g. missing 10-15% of doses) Definitions of Key Concepts in Phase I Trials
 Definitions of Key Concepts in Phase I Trials.")
11
Two examples from HCC trials Karim’s study: DLT will be defined as any one of the following attributed to AZD5363/AZD1208 during course 1: grade 4 neutropenia ≥ 1 week; febrile neutropenia; grade 4 thrombocytopenia; grade 3 thrombocytopenia with bleeding; QTcF prolongation to > 500 msec, or an increase of > 60 msec from baseline QTcF to a QTcF value > 480 msec, confirmed on repeat EKG; grade 3 or 4 nausea, vomiting, or diarrhea despite use of adequate medical intervention; any other clinically significant ≥ grade 3 non-hematologic toxicity; and/or persistent, intolerable toxicity which delays scheduled treatment for > 14 days. All subjects will be evaluated for toxicity by history, physical exam, and clinical labs weekly during course 1, every 4 weeks during subsequent courses, and at EOS. Subjects will undergo imaging every 2 courses (approximately 8 weeks), and will remain on study as long as they are deriving clinical benefit, tolerating the combination, and consent to continue. Blood samples will be drawn at frequent intervals during course 1 to assess the pharmacokinetic behavior of AZD5363 + AZD1208. Disease-specific cancer markers, AKT-regulated biomarkers, and PIM-regulated biomarkers will be evaluated pre-and post-treatment. One specific toxicity concern is hyperglycemia, which has been noted with both agents. Adoptive T-cell transfer study: The DLT in our studies will be identical to DLT defined in NCI Surgery Branch adoptive T cell transfer protocols, which is 1) ≥Grade 2 or more bronchospasm or generalized uticaria (hypersensitivity), 2) ≥Grade 2 or more allergic reactions, 3) Grade 3 or greater toxicity occurring within 24 hours post cell infusion (related to cell infusion) and not reversible to a grade 2 or less within 8 hours with two doses of 650 mg po of acetaminophen or two doses of 50 mg po of dyphenhydramine, 4) any Grade 4 autoimmunity, 5) Grade 3 autoimmunity (excluding vitiligo, the depigmentation of the skin and/or hair) that cannot be resolved to less than or equal to a grade 2 autoimmune toxicity within 10 days, or 4) any grade 3 or greater non-hematologic toxicity, excluding injection site reactions, skin rash, pruritis, and local adenopathy (Severe skin rashes such as Steven-Johnson Syndrome or TEN will be considered DLT), 6) > Grade 3 toxicity of any kind related to the TIL 1383I TCR transduced T cells administration with particular attention to the following events: a) ≥Grade 3 injection site reaction due to TIL 1383I TCR transduced T cells administration, b) > Grade 3 hematological or hepatic toxicity that does not subside within 4 weeks after infusion of TIL 1383I TCR transduced T cells, c) > Grade 3 neurotoxicity: New motor or sensory deficits, encephalopathy, or signs and symptoms that may indicate either tumor progression or a productive immune response will be evaluated as noted in earlier in this section with radiological imaging, and, if warranted, biopsy.
, and will remain on study as long as they are deriving clinical benefit, tolerating the combination, and consent to continue. Blood samples will be drawn at frequent intervals during course 1 to assess the pharmacokinetic behavior of AZD AZD1208. Disease-specific cancer markers, AKT-regulated biomarkers, and PIM-regulated biomarkers will be evaluated pre-and post-treatment. One specific toxicity concern is hyperglycemia, which has been noted with both agents. Adoptive T-cell transfer study: The DLT in our studies will be identical to DLT defined in NCI Surgery Branch adoptive T cell transfer protocols, which is 1) ≥Grade 2 or more bronchospasm or generalized uticaria (hypersensitivity), 2) ≥Grade 2 or more allergic reactions, 3) Grade 3 or greater toxicity occurring within 24 hours post cell infusion (related to cell infusion) and not reversible to a grade 2 or less within 8 hours with two doses of 650 mg po of acetaminophen or two doses of 50 mg po of dyphenhydramine, 4) any Grade 4 autoimmunity, 5) Grade 3 autoimmunity (excluding vitiligo, the depigmentation of the skin and/or hair) that cannot be resolved to less than or equal to a grade 2 autoimmune toxicity within 10 days, or 4) any grade 3 or greater non-hematologic toxicity, excluding injection site reactions, skin rash, pruritis, and local adenopathy (Severe skin rashes such as Steven-Johnson Syndrome or TEN will be considered DLT), 6) > Grade 3 toxicity of any kind related to the TIL 1383I TCR transduced T cells administration with particular attention to the following events: a) ≥Grade 3 injection site reaction due to TIL 1383I TCR transduced T cells administration, b) > Grade 3 hematological or hepatic toxicity that does not subside within 4 weeks after infusion of TIL 1383I TCR transduced T cells, c) > Grade 3 neurotoxicity: New motor or sensory deficits, encephalopathy, or signs and symptoms that may indicate either tumor progression or a productive immune response will be evaluated as noted in earlier in this section with radiological imaging, and, if warranted, biopsy..")
12
Maximum Administered Dose (MAD), Maximum Tolerated Dose (MTD): confusing ◦ Usage of these 2 phrases varies with country More important term: Recommended phase II dose (RPTD or RD): ◦ Dose associated with DLT in a pre-specified proportion of patients; dose that will be used in subsequent phase II trials ◦ Often the dose that is selected before launching to phase II is based on much more than just DLTs in cycle 1 ◦ Cycle 2 and beyond DLTs and other AEs ◦ PK profile ◦ Clinical outcomes ◦ Immune responses ◦ Aside: phase I/II study ◦ What is gained and what is lost by combining two phases in one protocol? Definitions of Key Concepts in Phase I Trials

13
Phase I Trial Design: Classic Algorithmic Approach “Standard” Phase I trials (in oncology) use what is often called the ‘3+3’ design (aka ‘modified Fibonacci’): Maximum tolerated dose (MTD) is considered highest dose at which 1 or 0 out of six patients experiences DLT. Doses need to be pre-specified Confidence in MTD is usually poor. Treat 3 patients at dose K 1.If 0 patients experience dose-limiting toxicity (DLT), escalate to dose K+1 2.If 2 or more patients experience DLT, de-escalate to level K-1 3.If 1 patient experiences DLT, treat 3 more patients at dose level K A.If 1 of 6 experiences DLT, escalate to dose level K+1 B.If 2 or more of 6 experiences DLT, de-escalate to level K-1
, escalate to dose K+1 2.If 2 or more patients experience DLT, de-escalate to level K-1 3.If 1 patient experiences DLT, treat 3 more patients at dose level K A.If 1 of 6 experiences DLT, escalate to dose level K+1 B.If 2 or more of 6 experiences DLT, de-escalate to level K-1.")
14
Attributed to a merchant from the 13th century ◦ Mathematical Model having nothing specifically to do with drug development. Sounds fancy, but no real basis. Doses increase by: 100%, 66%, 50%, 40%, 33%, etc. Standard “3+3” design: 3 patients per cohort, escalating to 6 if DLT occurs Dose escalate until DLT observed and MTD/RP2D defined Advantages: ◦ relatively safe, straightforward, clinician-friendly Disadvantages: ◦ lacks statistical foundation and precision, potentially treating a large proportion of patients at sub-therapeutic doses, time consuming ◦ Low confidence in selected dose as ‘safe’ or even near the desired DLT rate Phase I Trial Design: “Modified Fibonacci “ Dose Escalation (Algorithmic)
.")
15
3 pts Dose 3 pts Recommended dose (some call this MTD in US) DLT 3 pts+ DLT MAD Phase I Trial Design: Standard 3 + 3 Design
 DLT 3 pts+ DLT MAD Phase I Trial Design: Standard Design")
16
Optimal biological dose (OBD): ◦ Dose associated with a pre-specified desired effect on a biomarker ◦ Examples: ◦ Dose at which > XX% of patients have inhibition of a key target in tumor/surrogate tissues ◦ Dose at which > XX% of patients achieve a pre-specified immunologic parameter ◦ Challenge with defining OBD is that the “desired effect on a biomarker” is generally not known or validated before initiation of the phase I trial Definitions of Key Concepts in Phase I Trials
: ◦ Dose associated with a pre-specified desired effect on a biomarker ◦ Examples: ◦ Dose at which > XX% of patients have inhibition of a key target in tumor/surrogate tissues ◦ Dose at which > XX% of patients achieve a pre-specified immunologic parameter ◦ Challenge with defining OBD is that the desired effect on a biomarker is generally not known or validated before initiation of the phase I trial Definitions of Key Concepts in Phase I Trials")
17
Pharmacokinetics (PK): ◦ “what the body does to the drug” ◦ absorption, distribution, metabolism and excretion ◦ PK parameters: Cmax, AUC (drug exposure), t1/2, Clearance, etc. Pharmacodynamics (PD): ◦ “what the drug does to the body” ◦ e.g. nadir counts, non-hematologic toxicity, molecular correlates, imaging endpoints Definitions of Key Concepts in Phase I Trials
: ◦ what the drug does to the body ◦ e.g. nadir counts, non-hematologic toxicity, molecular correlates, imaging endpoints Definitions of Key Concepts in Phase I Trials.")
18
At what dose do you start? What type of patients? How many patients per cohort? How quickly do you escalate? What are the endpoints? Phase I Trials: Fundamental Questions

19
At what dose do you start? What type of patients? How many patients per cohort? How (quickly) do you escalate? What are the endpoints? Phase I Trials: Fundamental Questions
 do you escalate. What are the endpoints. Phase I Trials: Fundamental Questions.")
20
Typically a rodent (mouse or rat) and non-rodent (dog or non-human primate) species Reality of animal organ specific toxicities – very few predict for human toxicity ◦ Myelosuppression and GI toxicity more predictable ◦ Hepatic and renal toxicities – large false positive Toxicologic parameters: ◦ LD 10 – lethal dose in 10% of animals ◦ TDL (toxic dose low) – lowest dose that causes any toxicity in animals ◦ NOAEL – no observed adverse effect level Preclinical Toxicology
 and non-rodent (dog or non-human primate) species Reality of animal organ specific toxicities – very few predict for human toxicity ◦ Myelosuppression and GI toxicity more predictable ◦ Hepatic and renal toxicities – large false positive Toxicologic parameters: ◦ LD 10 – lethal dose in 10% of animals ◦ TDL (toxic dose low) – lowest dose that causes any toxicity in animals ◦ NOAEL – no observed adverse effect level Preclinical Toxicology")
21
1/10 of the LD10 in rodents or (depending on sensitivity of the species) 1/6 or 1/3 of the TDL in large animals Unless preclinical studies suggest a very steep dose/toxicity curve Phase I Trials: Starting Dose
 1/6 or 1/3 of the TDL in large animals Unless preclinical studies suggest a very steep dose/toxicity curve Phase I Trials: Starting Dose")
22
At what dose do you start? What type of patients? How many patients per cohort? How (quickly) do you escalate? What are the endpoints? Phase I Trials: Fundamental Questions
 do you escalate. What are the endpoints. Phase I Trials: Fundamental Questions.")
23
“Conventional” eligibility criteria- examples: ◦ Advanced solid tumors (or select hematologic malignancies) unresponsive to standard therapies or for which there is no known effective treatment ◦ Performance status (e.g. ECOG 0 or 1) ◦ Adequate organ function (e.g. ANC, platelets, creatinine, AST/ALT, bilirubin) ◦ Specification about prior therapy allowed ◦ Specification about time interval between prior therapy and initiation of study treatment ◦ No serious uncontrolled medical disorder or active infection Phase I Patient Population
 ◦ Adequate organ function (e.g. ANC, platelets, creatinine, AST/ALT, bilirubin) ◦ Specification about prior therapy allowed ◦ Specification about time interval between prior therapy and initiation of study treatment ◦ No serious uncontrolled medical disorder or active infection Phase I Patient Population.")
24
“Agent-specific” eligibility criteria - examples: ◦ Restriction to certain patient populations – must have strong scientific rationale ◦ Specific organ functions: ◦ Cardiac function (e.g. QTc 45%, etc) ◦ if preclinical or prior clinical data of similar agents suggest cardiac risks ◦ No recent (6-12 months) history of acute MI/unstable angina, cerebrovascular events, venous thromboembolism; no uncontrolled hypertension; no significant proteinuria (eg. antiangiogenic agents) ◦ Prohibited medications if significant risk of drug interaction How has this occurred in the era of immunotherapies (e.g. PD-1/PD-L1 checkpoint blockade?) Phase I Patient Population
 ◦ if preclinical or prior clinical data of similar agents suggest cardiac risks ◦ No recent (6-12 months) history of acute MI/unstable angina, cerebrovascular events, venous thromboembolism; no uncontrolled hypertension; no significant proteinuria (eg. antiangiogenic agents) ◦ Prohibited medications if significant risk of drug interaction How has this occurred in the era of immunotherapies (e.g. PD-1/PD-L1 checkpoint blockade ) Phase I Patient Population.")
25
At what dose do you start? What type of patients? How many patients per cohort? How (quickly) do you escalate? What are the endpoints? Phase I Trials: Fundamental Questions
 do you escalate. What are the endpoints. Phase I Trials: Fundamental Questions.")
26
Number of Patients per Cohort Predicated on several factors ◦What is driving the definition of ‘optimal dose’? That is, what is the primary endpoint? ◦For toxicity-driven designs ◦Usually 3 per cohort for 3+3 and even model-based. ◦Accelerated Titration Design attempts to minimize Pt # ◦Typically 1 patient per cohort until >grade 2 toxicity and then increase to 3 (or 6 if DLT) ◦For efficacy-driven designs ◦Usually more than 3 per cohort, but trial-dependent.
 ◦For efficacy-driven designs ◦Usually more than 3 per cohort, but trial-dependent..")
27
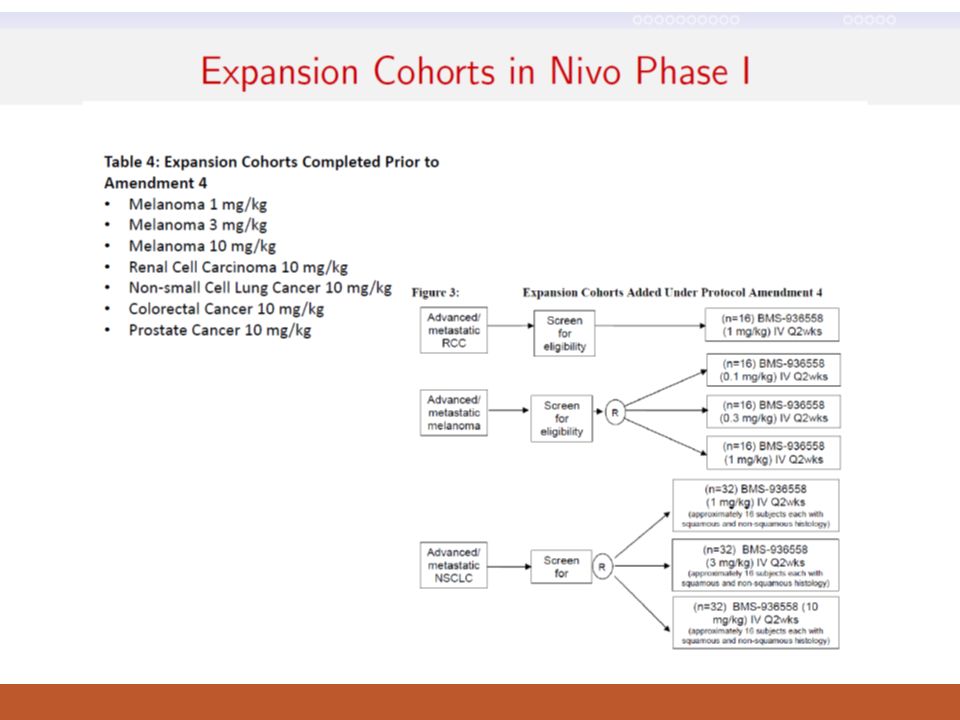
Many phase I trials accrue additional patients at the RP2D to obtain more safety, PK, PD data (but this expansion cohort does not equal to a phase II trial) “EXPANSION COHORT” Cohort Dose Escalation: Standard 3 + 3 Design
 EXPANSION COHORT Cohort Dose Escalation: Standard Design")
28
At what dose do you start? What type of patients? How many patients per cohort? How (quickly) do you escalate? What are the endpoints? Phase I Trials: Fundamental Questions
 do you escalate. What are the endpoints. Phase I Trials: Fundamental Questions.")
29
Start with a safe starting dose ◦ Often you start with ‘dose level 2’ and define dose level 1 as a ‘step-down dose’ if the starting dose is too toxic. Easier than amending the trial to add a lower level. Minimize the number of pts treated at sub-toxic (and thus maybe sub- therapeutic) doses Escalate dose rapidly in the absence of toxicity Escalate dose slowly in the presence of toxicity Different therapies have difference ‘increments’ ◦ Standard chemo: you might double the dose for the next dose level ◦ Adoptive T-cell transfer: you might increase dose by factor of 10. (e.g. dose level 1 = 1x10 7 ; dose level 2 = 1x10 8 ) Phase I Trial Basic Principles
 doses Escalate dose rapidly in the absence of toxicity Escalate dose slowly in the presence of toxicity Different therapies have difference ‘increments’ ◦ Standard chemo: you might double the dose for the next dose level ◦ Adoptive T-cell transfer: you might increase dose by factor of 10. (e.g. dose level 1 = 1x10 7 ; dose level 2 = 1x10 8 ) Phase I Trial Basic Principles.")
30
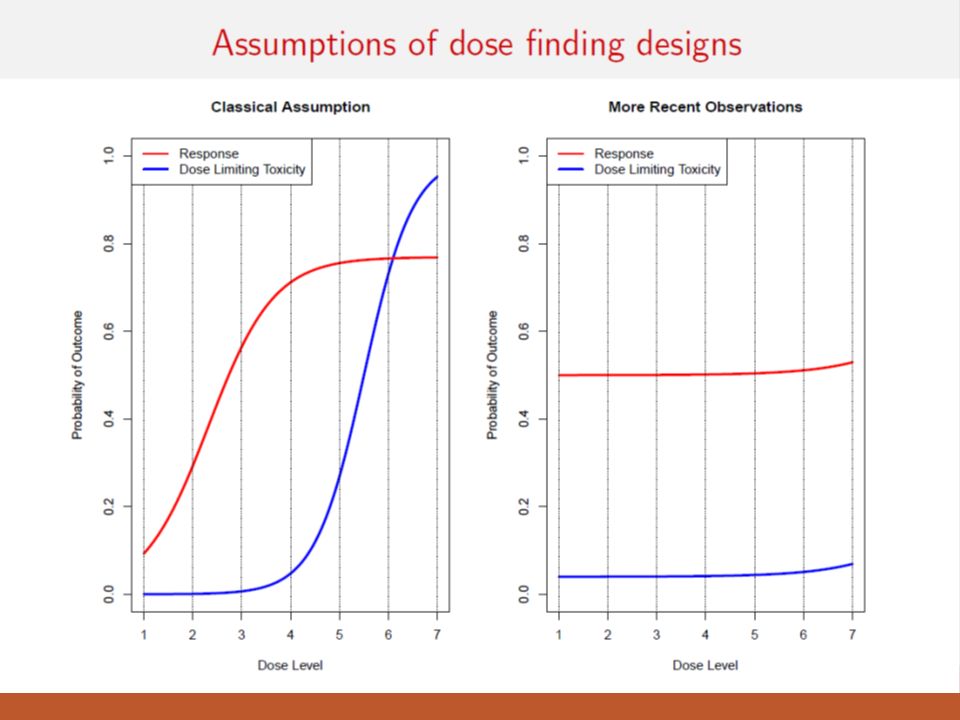
The higher the dose, the greater the likelihood of efficacy ◦ Dose-related acute toxicity regarded as a surrogate for efficacy ◦ Highest safe dose is dose most likely to be efficacious ◦ This dose-effect assumption is primarily for cytotoxic agents and may not apply to molecularly targeted agents Modern situations where this isn’t true: ◦ Nivo and Pembro: Pretty flat dose response curves ◦ Temsirolimus in kidney cancer (Atkins et al., JCO, 2004) ◦ Anastrazole in breast cancer (Jonat et al., Eur J Cancer, 1996) “Old School” Phase I Trial Assumption
 ◦ Anastrazole in breast cancer (Jonat et al., Eur J Cancer, 1996) Old School Phase I Trial Assumption")
32
First proposed by Simon et al (J Natl Cancer Inst 1997) Several variations exist: ◦ Doubling dose in single-patient cohorts till Grade 2 toxicity ◦ then revert to standard 3+3 design using a 40% dose escalation ◦ Modifications are often trial/drug dependent ◦ Intra-patient dose escalation allowed in some variations Advantage ◦ More rapid dose escalation ◦ Fewer patient numbers ◦ Typically does not expedite trial completion Phase I Trial Design: Accelerated Titration Design (Rule-based)
 Several variations exist: ◦ Doubling dose in single-patient cohorts till Grade 2 toxicity ◦ then revert to standard 3+3 design using a 40% dose escalation ◦ Modifications are often trial/drug dependent ◦ Intra-patient dose escalation allowed in some variations Advantage ◦ More rapid dose escalation ◦ Fewer patient numbers ◦ Typically does not expedite trial completion Phase I Trial Design: Accelerated Titration Design (Rule-based)")
33
1 pt Dose 1 pt 3 pts Recommended dose DLT Starting dose 3 pts+ DLT Gr 2 toxicity Phase I Trial Design: Accelerated Titration

34
Combination phase I trials: ◦ New drug A + Standard drug B ◦ Need to provide rationale: why add A to B? ◦ Need to think about overlapping toxicity in your definition of DLT ◦ Ideally keep standard drug dose fixed and escalate the new drug (e.g. 1/2, 2/3, full dose) Phase I Trial of Combination of Agents
 Phase I Trial of Combination of Agents.")
35
DL 123451234 123451234 Relative dose AB DC Phase I Trial of Combinations of Agents

36
Chronic and cumulative toxicities usually cannot be assessed & may be missed ◦ Most patients do not stay on trial beyond 2 cycles Uncommon toxicities will be missed ◦ Too few patient numbers in a phase I trial ◦ Reason for toxicity evaluation & reporting through phase IV drug testing Pitfalls of Phase I Trials

37
At what dose do you start? What type of patients? How many patients per cohort? How quickly do you escalate? What are the endpoints? Phase I Trials: Fundamental Questions

38
DLT and other toxicity – safety and tolerability Pharmacokinetics (PK) Pharmacodynamics (PD) ◦ biological correlates, imaging endpoints Preliminary antitumor activity Expansion cohort: Have become THE NORM in phase I studies. ◦ Historically: treat a few more patients at MTD to ‘confirm’ safety, etc. ◦ More recently: ‘phase II’-like but without clear objectives and endpoints (NB: statisticians do not like unclear objectives!). Endpoints in Phase I Trials
. Endpoints in Phase I Trials.")
39
TrialNo. of Trials No. of Patients Assessed for Response Overall Response Rate* % No. of Patients Assessed for Toxic Events Deaths from Toxic Events no. % Total First use of an agent in humans 11731644.834989 (0.26%) Cytotoxic chemotherapy First use of an agent in humans 4312985.014227 (0.49%) Immunomodulator First use of an agent in humans 164047.44311 (0.23%) Receptor or signal transduction First use of an agent in humans 277423.88531 (0.12%) Antiangiogenesis First use of an agent in humans 82007.02280 Gene transfer First use of an agent in humans 00000 Vaccine First use of an agent in humans 235203.15640 Response Rates and Deaths from Toxic Events in Phase I Oncology Trials Involving the First Use of Agents in Humans (Horstman et al, NEJM 352, 2005)
 Cytotoxic chemotherapy First use of an agent in humans (0.49%) Immunomodulator First use of an agent in humans (0.23%) Receptor or signal transduction First use of an agent in humans (0.12%) Antiangiogenesis First use of an agent in humans Gene transfer First use of an agent in humans Vaccine First use of an agent in humans Response Rates and Deaths from Toxic Events in Phase I Oncology Trials Involving the First Use of Agents in Humans (Horstman et al, NEJM 352, 2005).")
40
Patient Pre-selection ◦ Other than typical Inclusion/Exclusion Criteria ◦ Tumor type ◦ Eg. Gorlin’s syndrome, basal cell carcinoma – HH inhibitors ◦ Molecular Profiling ◦ bRAF melanoma ◦ ALK+/EGFR+ NSCLCa ◦ What about other targets? Clinical Development of Molecularly Targeted Agents: Known Challenges

41
General requirement for long-term administration: pharmacology and formulation critical Difficulty in determining the optimal dose in phase I: MTD versus OBD ◦ Do we need MTD with targeted agents? Absent or low-level tumor regression as single agents: problematic for making go no-go decisions Need for large randomized trials to definitively assess clinical benefit: need to maximize chance of success in phase III Clinical Development of Molecularly Targeted Agents: Known Challenges

42
Conventional cytotoxic drugs have led to predictable effects on proliferating tissues (neutropenia, mucositis, diarrhea), thus enabling dose selection and confirming mechanisms of action Targeted biological agents may or may not have predictable effects on normal tissues and often enter the clinic needing evidence/proof of mechanisms in patients ◦ On target vs off-target effects Biological Correlative Studies: Why Do We Need Them?
, thus enabling dose selection and confirming mechanisms of action Targeted biological agents may or may not have predictable effects on normal tissues and often enter the clinic needing evidence/proof of mechanisms in patients ◦ On target vs off-target effects Biological Correlative Studies: Why Do We Need Them")
43
Prognostic Markers Correlate with disease outcome regardless of intervention e.g. Clinical: stage, PS e.g. ER in breast cancer e.g. HPV in H&N cancer Predictive Markers Predict outcome with specific therapy - match drugs with appropriate pts e.g. KRAS mutations and anti- EGFR monoclonal antibodies in colorectal cancer e.g. HER2 in breast cancer Pharmacodynamic Markers Confirm biological activity e.g. pERK with a targeted agent (such as a MEK inhibitor) Biomarkers: Functional Definitions
 Biomarkers: Functional Definitions.")
44
Should be driven by sound scientific rationale Phase I: ◦ Proof-of-mechanism ◦ Exploratory ◦ Establish optimal biological dose and/or schedule in some trials (especially if little or no toxicity expected) ◦ Often more practical to perform at an expanded cohort at the recommended phase II dose Use of Lab Correlates in Clinical Trials
 ◦ Often more practical to perform at an expanded cohort at the recommended phase II dose Use of Lab Correlates in Clinical Trials")
45
PAR Levels in Tumors of Predose vs. Postdose NCTVL

46
Pharmaceutical industries / biotechnology companies (big and small) ◦ Big pharmas: often select “preferred sites” for pipeline development, often intense “test burden”, secure and well funded ◦ Small pharmas: 1 or 2 drugs as their “life-line”, more amenable to data sharing, less secure Academic agencies (NCI US, EORTC, etc) In-house development (e.g. Apogee phase I study @ HCC) Challenges: ◦ Getting support for investigator-initiated trial ideas ◦ Getting different agents from different companies for a single trial Sources of Phase I Drugs
 Challenges: ◦ Getting support for investigator-initiated trial ideas ◦ Getting different agents from different companies for a single trial Sources of Phase I Drugs.")
47
Investigators Trial nurses Fellows Data coordinators Pharmacists Radiologists Lab personnel: reference, PK, PD Biostatisticians Scientists PATIENT The Successful Phase I Team

48
Recent Changes

Similar presentations










 Polymerase (PARP) by ABT-888 in Patients With Advanced Malignancies: Results of a Phase 0 Trial Shivaani Kummar, MD National.>")





 Polymerase.>")









 clinical trials in healthy volunteer subjects Oncology Drug Advisory Committee.>")
 Dose Finding Studies. Dose Finding Dose finding trials: broad class of early development trial designs whose purpose is to find.>")

