Download presentation
Presentation is loading. Please wait.

1
Hemoglobin; structure and function Mahmoud A. Alfaqih BDS PhD Jordan University of Science and Technology (JUST)
.")
2
Heme-proteins Heme-proteins are a group of specialized proteins that contains heme (prosthetic group). Examples of hemeproteins: I- Myoglobin and hemoglobin II- Other Hemeproteins 1- Cytochromes 2- Peroxidases 3- catalase

3
MYOGLOBIN AND HEMOGLOBIN Both proteins are formed of heme attached to a globin chain Myoglobin is formed of one heme attached to one polypeptide chain, while Hemoglobin is formed of four heme groups attached to four polypeptide chains i.e. one heme per one chain.

4
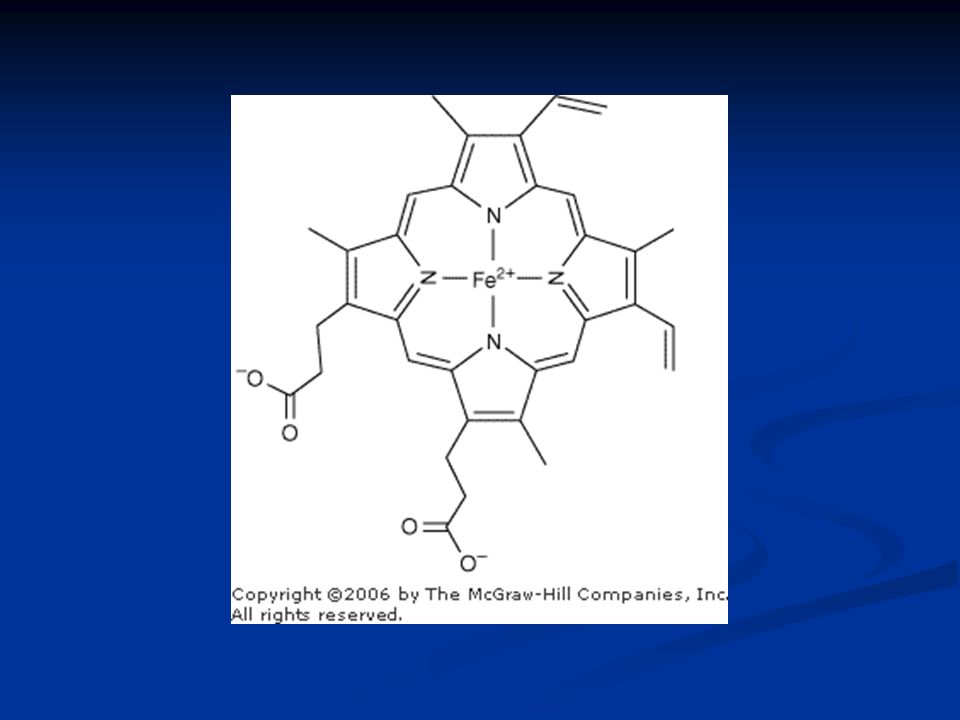
Heme A cyclic tetrapyrrole consisting of four molecules of pyrrole linked by methylene bridges A cyclic tetrapyrrole consisting of four molecules of pyrrole linked by methylene bridges This planar network of conjugated double bonds absorbs visible light and colors heme deep red The substituents at the -positions of heme are methyl (M), vinyl (V), and propionate (Pr) groups arranged in the order M, V, M, V, M, Pr, Pr, M One atom of ferrous iron (Fe 2 + ) resides at the center of the planar tetrapyrrole Oxidation of the Fe 2 + of myoglobin or hemoglobin to Fe 3 + destroys their biologic activity
, vinyl (V), and propionate (Pr) groups arranged in the order M, V, M, V, M, Pr, Pr, M One atom of ferrous iron (Fe 2 + ) resides at the center of the planar tetrapyrrole Oxidation of the Fe 2 + of myoglobin or hemoglobin to Fe 3 + destroys their biologic activity")
5
Heme is a complex of protoporphyrin IX and ferrous iron (Fe 2+ ). M = methyl V = Vinyl P = Propionyl

7
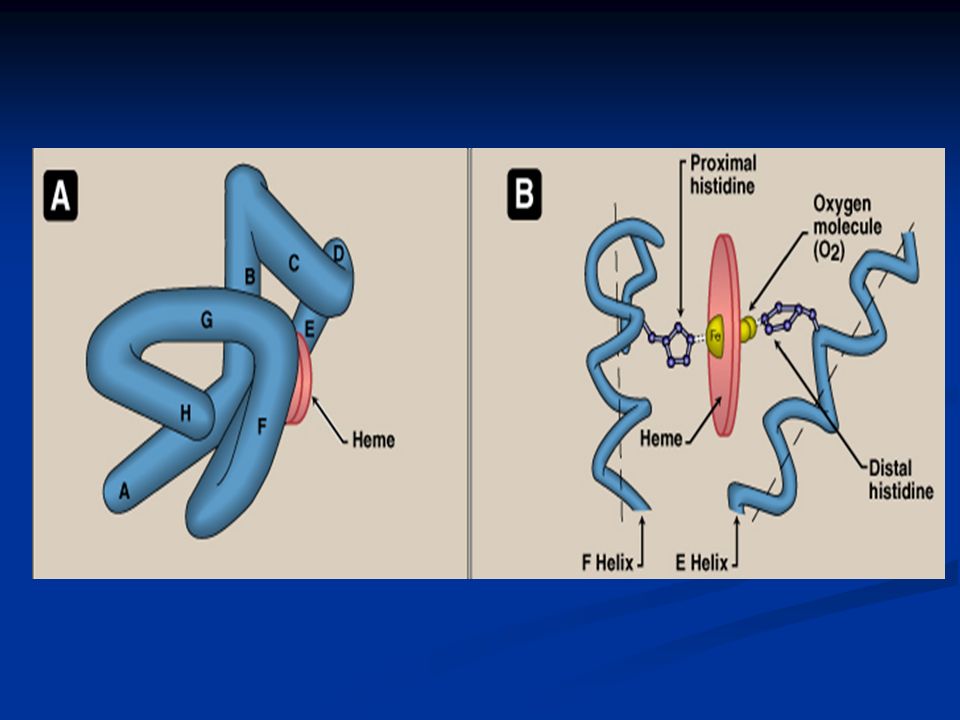
Attachment of Heme to The Polypeptide Chain Ferrous of heme can form up to 6 bonds as follows: 1- Four bonds are formed between Fe 2+ and the four nitrogens of the porphyrin ring. 2- The fifth bond is for histidine (His F8) of the globin chain. 3- The sixth bond is for oxygen which lies between Fe ++ and another histidine termed distal histidine (E7)
 of the globin chain. 3- The sixth bond is for oxygen which lies between Fe ++ and another histidine termed distal histidine (E7).")
8
A-Myoglobin Location: In skeletal and cardiac muscles Function: a storage form of oxygen in muscles, enhances transport of oxygen inside muscle cells Structure : single polypeptide chain, similar to individual subunit polypeptides of hemoglobin

9
Oxygen stored in red muscle myoglobin is released during O 2 deprivation (eg, severe exercise) for use in muscle mitochondria for aerobic synthesis of ATP
 for use in muscle mitochondria for aerobic synthesis of ATP")
10
Helical content of Myoglobin Single polypeptide chain, 80% helical, forms 8 helices named starting from the N-terminal A through H Soluble protein with polar outer surface and non- polar inner groups except for the two histidines that function in oxygen binding i.e. histidines F8 and E7.

12
Hemoglobin Location: Exclusively in red blood cells Function: Transports oxygen from lungs to every tissue in the body Removes CO 2 from tissues Also Hb / oxy-Hb system acts as buffer in RBCs

13
Note Myoglobin & the β Subunits of Hemoglobin Share Almost Identical Secondary and Tertiary Structures

14
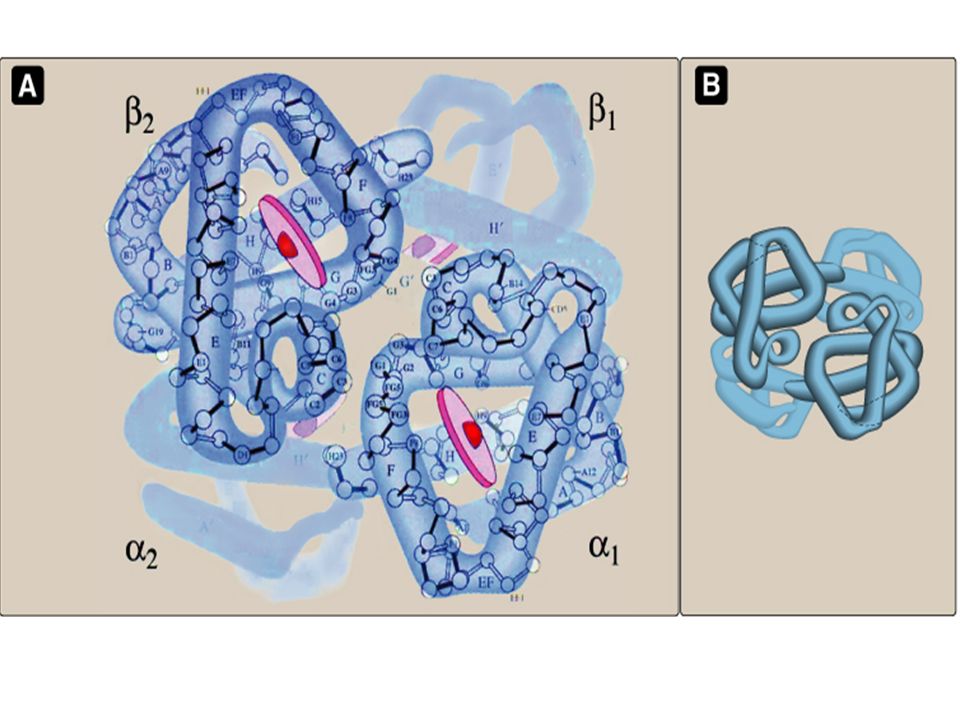
Structure Of Adult Hemoglobin

15
HbA is formed of four polypeptide chains Two hetero-dimers, dimer is formed of one alpha (α) and one beta (β) chain Tetrameric structure (αβ) 2 i.e. (α 1 β 1 – α 2 β 2 ) Each subunit is similar in structure to Myoglobin 1- HbA
 Each subunit is similar in structure to Myoglobin 1- HbA.")
16
Continued The α & β chains are tightly held together by hydrophobic interactions The two dimers are less tightly held together by hydrogen & ionic bonds Allows movement of the 2 dimers relative to each other. Movement occurs during oxygenation & deoxygenation

18
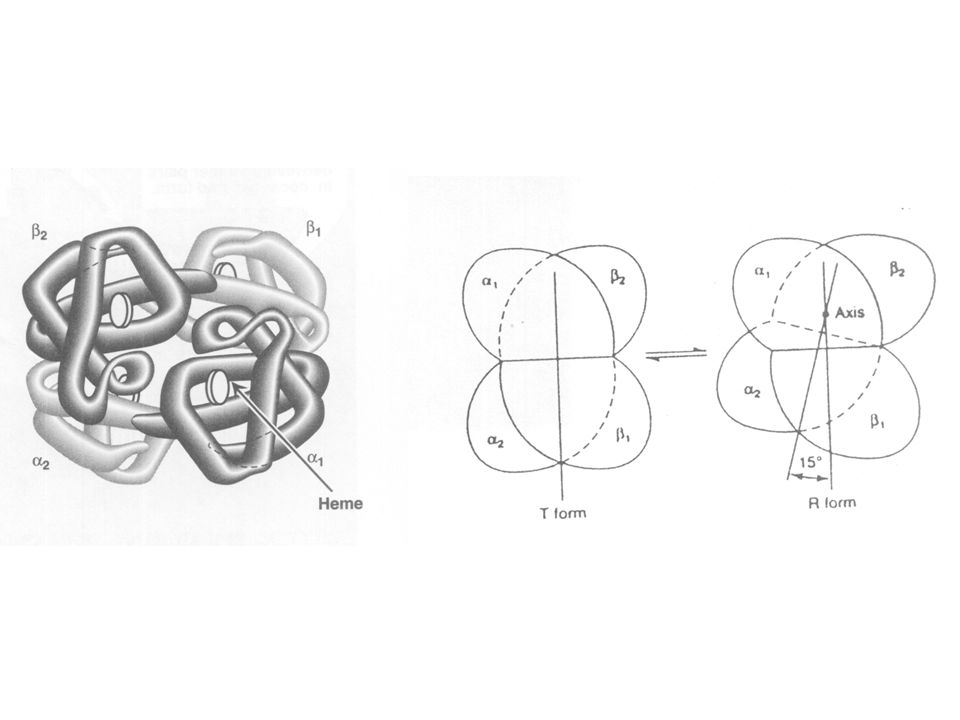
The “ T “ form ( Hb ): OR [ Taut ( tense ) Form ] Low-oxygen affinity form of hemoglobin Network of ionic bonds between the two dimers, constrains movement Dimers are difficult to move relative to each other
![The T form ( Hb ): OR [ Taut ( tense ) Form ] Low-oxygen affinity form of hemoglobin Network of ionic bonds between the two dimers, constrains movement Dimers are difficult to move relative to each other](http://images.slideplayer.com/36/10560101/slides/slide_18.jpg "The T form ( Hb ): OR [ Taut ( tense ) Form ] Low-oxygen affinity form of hemoglobin Network of ionic bonds between the two dimers, constrains movement Dimers are difficult to move relative to each other")
19
The “ R “ Form ( HbO2 ): OR [ Relaxed Form ] Binding of oxygen disrupts ionic and hydrogen bonds between two dimers Dimers are free to move relative to each other It has a higher affinity to oxygen.
![The R Form ( HbO2 ): OR [ Relaxed Form ] Binding of oxygen disrupts ionic and hydrogen bonds between two dimers Dimers are free to move relative to each other It has a higher affinity to oxygen.](http://images.slideplayer.com/36/10560101/slides/slide_19.jpg "The R Form ( HbO2 ): OR [ Relaxed Form ] Binding of oxygen disrupts ionic and hydrogen bonds between two dimers Dimers are free to move relative to each other It has a higher affinity to oxygen.")
22
Binding Of oxygen to myoglobin and hemoglobin Myoglobin can bind only one molecule of oxygen (O 2 ), because it contains only one heme group Hemoglobin can bind four oxygen molecules The degree of saturation (Y) can vary between zero (all sites are empty) and 100% (all sites are full)
, because it contains only one heme group Hemoglobin can bind four oxygen molecules The degree of saturation (Y) can vary between zero (all sites are empty) and 100% (all sites are full)")
23
Oxygen dissociation curve A plot of Y measured at different partial pressures of oxygen (pO 2 ) Myoglobin has a higher oxygen affinity at all pO 2 values (P 50 ) is 1 mm Hg for myoglobin and 26 mm Hg for hemoglobin.
 Myoglobin has a higher oxygen affinity at all pO 2 values (P 50 ) is 1 mm Hg for myoglobin and 26 mm Hg for hemoglobin.")
25
Oxygen dissociation curve of Myoglobin It has a hyperbolic shape Oxygenated (MbO 2 ) and deoxygenated (Mb) myoglobin exist in a simple equilibrium: :
 and deoxygenated (Mb) myoglobin exist in a simple equilibrium: :")
26
Oxygen dissociation curve of Hemoglobin Sigmoidal in shape Subunits cooperate in binding oxygen Binding of an oxygen molecule at one heme group increases the affinity of the remaining groups to O 2 This is referred to as heme-heme interaction :

27
Hemoglobin binds oxygen with increasing affinity It is more difficult for the first oxygen molecule to bind to hemoglobin, subsequent binding of oxygen occurs with higher affinity Affinity of hemoglobin for the last oxygen bound is approximately 300 times greater than its affinity for the first oxygen bound

28
Allosteric effects Interaction at one site on the hemoglobin molecule affects the binding of oxygen to heme groups at other locations on the molecule Allosteric effectors: 1. pO 2 2. pH 3. pCO 2 4. 2,3 - bisphosphoglycerate

29
Bohr effect Affinity of hemoglobin for oxygen decreases when the pH is lowered (when the partial pressure of CO 2 Affinity of hemoglobin for oxygen decreases when the pH is lowered (when the partial pressure of CO 2 increases) Oxygen dissociation curve shifts to the right Stabilization of the T state of Hemoglobin
 Oxygen dissociation curve shifts to the right Stabilization of the T state of Hemoglobin")
30
Bohr effect

31
Source of Protons The concentration of CO 2 and H + is higher around metabolically active tissues compared to lung alveoli CO 2 is converted by carbonic anhydrase to carbonic acid: Carbonic acid spontaneously loses a proton: Lungs have higher pH compared to tissues : ):
:")
32
Source of protons…..cont. Hemoglobin carries CO 2 as carbamates formed with the amino terminal nitrogens of the polypeptide chains Formation of carbamates gets rid of the positive charge of terminal nitrogen and allows the gain of a net negative charge The above reaction also causes the release of protons

33
Source of protons The net negative charge gained following carbon dioxide binding favors salt bond formation between the α and β chains This change stabilizes T state and facilitates delivery of oxygen

34
What happens at the lung side? In the lungs, the process reverses. O 2 binds to deoxyhemoglobin. Protons are released and combine with bicarbonate to form carbonic acid. Dehydration of H 2 CO 3, catalyzed by carbonic anhydrase, forms CO 2, which is exhaled. Binding of oxygen drives the exhalation of CO 2

36
Mechanism of Bohr effect Deoxy form of hemoglobin has a greater affinity for protons than does oxyhemoglobin N-terminal α-amino groups and specific histidine side chains have higher pK a s in deoxyhemoglobin than in oxyhemoglobin A decrease in pH causes them to become protonated (charged) and form ionic bonds This effect stabilizes the deoxy form of hemoglobin
 and form ionic bonds This effect stabilizes the deoxy form of hemoglobin")
37
Continued An increase in protons shifts the equilibrium to the right (favoring deoxyhemoglobin) An increase in pO 2 (or a decrease in protons) shifts the equilibrium to the left
 An increase in pO 2 (or a decrease in protons) shifts the equilibrium to the left")
38
Tissue side

39
Lung side

40
Effect of 2,3-bisphosphoglycerate (BPG) on oxygen affinity It is the most abundant organic phosphate in the red blood cell BPG is synthesized from an intermediate of the glycolytic pathway
 on oxygen affinity It is the most abundant organic phosphate in the red blood cell BPG is synthesized from an intermediate of the glycolytic pathway")
41
Binding of 2,3-BPG to deoxyhemoglobin BPG decreases the oxygen affinity of hemoglobin by binding to deoxyhemoglobin but not to oxyhemoglobin This preferential binding stabilizes the taut conformation of deoxyhemoglobin

42
Binding site of 2,3-BPG BPG binds to a pocket, formed by the two β-globin chains, in the center of the deoxyhemoglobin tetramer This pocket contains several positively charged amino acids that form ionic bonds with BPG

43
Effect of 2,3-BPG on oxygen dissociation curve Hemoglobin from which 2,3- BPG has been removed has a high affinity for oxygen The presence of 2,3-BPG shifts the oxygen-dissociation curve to the right

44
Response of 2,3-BPG levels to chronic hypoxia or anemia The concentration of 2,3-BPG increases in chronic hypoxia, pulmonary emphysema, at high altitudes and in chronic anemia Elevated 2,3-BPG levels lower the oxygen affinity of hemoglobin, permitting greater unloading of oxygen to tissues

45
Adaptation to high altitude Physiologic changes due to prolonged exposure to high altitude include: Increase in the number of red cells Increase in the concentration of Hb Increase in the concentration of BPG

46
Role of 2,3-BPG in transfused blood Storing blood in acid-citrate-dextrose leads to a decrease of 2,3-BPG in RBCs Such blood fails to unload oxygen in the tissues Hemoglobin deficient in 2,3-BPG acts as an oxygen “trap” Transfused RBCs can restore 2,3-BPG in 24 to 48 hours This might represent a problem for the severely ill patient

47
Solution The decrease in 2,3-BPG can be prevented by adding inosine (hypoxanthine-ribose) Inosine can enter RBC, its ribose moiety is released and phosphorylated It then enters the hexose monophosphate pathway to be converted to 2,3-BPG
 Inosine can enter RBC, its ribose moiety is released and phosphorylated It then enters the hexose monophosphate pathway to be converted to 2,3-BPG")
48
Binding of CO 2 Most of the CO 2 is hydrated and transported as bicarbonate ion Some CO 2 is carried as carbamate bound to α-amino groups of hemoglobin (carbamino-hemoglobin) The binding of CO 2 stabilizes the T (taut) or deoxy form of (lower affinity for oxygen) The binding of CO 2 stabilizes the T (taut) or deoxy form of hemoglobin (lower affinity for oxygen)
 The binding of CO 2 stabilizes the T (taut) or deoxy form of (lower affinity for oxygen) The binding of CO 2 stabilizes the T (taut) or deoxy form of hemoglobin (lower affinity for oxygen)")
49
Binding of CO CO binds tightly to the hemoglobin iron When CO binds, hemoglobin shifts to the relaxed conformation This causes hemoglobin to bind oxygen with higher affinity This shifts curve to the left, and changes the normal shape to a hyperbola Hemoglobin is unable to release oxygen to the tissues

50
Effect of CO

51
HemoglobinMyoglobin Location In Red Blood CellsIn Cardiac & Skeletal Muscles No Of Polypeptides 4 polypeptidesOne polypeptide No Of Heme / molecule 4 HemeOne Heme No Of O 2 mol. Can bind 4 O 2 mol.One O 2 mol. Structure QuaternaryTertiary Affinity For O 2 ( At low pO 2 ) Lower than MyoglobinHigher than Hb O 2 Dissociation Curve Sigmoidal Cooperative Binding Of O 2 Hyperbolic Mb + O 2 MbO 2 Forms 2 forms R & TOne form Allosteric Effect AffectedNot
 Lower than MyoglobinHigher than Hb O 2 Dissociation Curve Sigmoidal Cooperative Binding Of O 2 Hyperbolic Mb + O 2 MbO 2 Forms 2 forms R & TOne form Allosteric Effect AffectedNot.")
52
Minor Hemoglobins Hb A is just one member of a family of proteins, the hemoglobins All hemoglobins are tetramers of two α-globin like chains and two β-globin- like chains

53
Fetal Hemoglobin (Hb F) A tetramer consisting of two α chains identical to those found in Hb A, plus two γ chains γ chains are members of the β-globin gene family Hb F's binds only weakly to BPG, it has a higher affinity for oxygen than Hb A
 A tetramer consisting of two α chains identical to those found in Hb A, plus two γ chains γ chains are members of the β-globin gene family Hb F s binds only weakly to BPG, it has a higher affinity for oxygen than Hb A")
54
Hb F synthesis during development Embryonic hemoglobin ( (ζ 2 ε 2 ), is synthesized in the first three months of pregnancy Hb F is the major hemoglobin in the fetus and newborn Hb F makes sixty percent of the total hemoglobin during the last months of fetal life Hb A synthesis starts at about the eighth month of pregnancy and gradually replaces Hb F.
, is synthesized in the first three months of pregnancy Hb F is the major hemoglobin in the fetus and newborn Hb F makes sixty percent of the total hemoglobin during the last months of fetal life Hb A synthesis starts at about the eighth month of pregnancy and gradually replaces Hb F.")
55
Hemoglobinpathies A family of genetic disorders caused by 1. production of structurally abnormal hemoglobin 2. synthesis of insufficient quantities of normal hemoglobin

56
Hemoglobinpathies Hemoglobinpathies include the following 1. Sickle cell anemia (Hb S) 2. Hemoglobin C disease (Hb C) 3. Thalassemia syndromes
 2. Hemoglobin C disease (Hb C) 3. Thalassemia syndromes.")
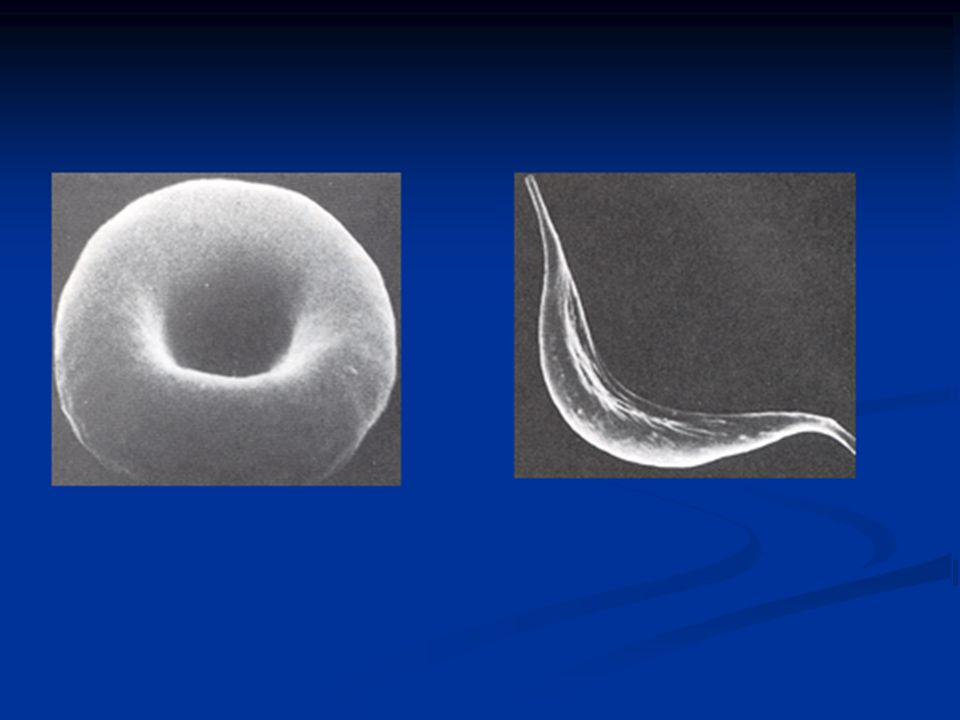
57
Sickle cell disease (Hemoglobin S disease) A genetic disorder caused by a single nucleotide alteration in the β-globin gene Sickle cell disease is a homozygous recessive disorder It occurs in individuals who have inherited two mutant genes Heterozygotes have one normal and one sickle cell gene Heterozygotes have sickle cell trait and can have a normal life span
 A genetic disorder caused by a single nucleotide alteration in the β-globin gene Sickle cell disease is a homozygous recessive disorder It occurs in individuals who have inherited two mutant genes Heterozygotes have one normal and one sickle cell gene Heterozygotes have sickle cell trait and can have a normal life span")
58
Amino acid substitution in Hb S β chains A molecule of Hb S contains two normal α-glob in chains and two mutant β-globin chains Glutamate at position six is replaced with valine

59
Effect of amino acid substitution The substitution forms a protrusion on the β-globin chain of hemoglobin Protrusion fits into a complementary site on the α chain of another hemoglobin molecule At low oxygen, deoxyhemoglobin S polymerizes into a network of fibres that distort RBCs sickled cells block the flow of blood in capillaries. This leads to localized anoxia (oxygen deprivation) in the tissueand eventually death (infarction) of tissue cells
 in the tissueand eventually death (infarction) of tissue cells.")
60
Variables that increase sickling 1.Decrease in pO 2 2.Increase in pCO 2 3.Increase in BPG concentration 4.Decrease in pH

62
Hemoglobin C disease A hemoglobin variant that has a single amino acid substitution in the sixth position of β-globin A lysine is substituted for glutamate Patients homozygous for hemoglobin C have a mild, chronic hemolytic anemia No specific therapy is required for these patients

63
Hemoglobin SC disease Some β-globin chains have the sickle cell mutation Other β-globin chains have Hemoglobin C mutation Hemoglobin levels tend to be higher in Hb SC disease than in sickle cell disease Patients with Hb SC disease remain well, until they suffer an infarctive crisis This crisis may follow childbirth or surgery and may be fatal

64
Hemoglobin electrophoresis Some β-globin chains have the sickle cell mutation Other β-globin chains have Hemoglobin C mutation Hemoglobin levels tend to be higher in Hb SC disease than in sickle cell disease Patients with Hb SC disease remain well, until they suffer an infarctive crisis This crisis may follow childbirth or surgery and may be fatal

65
Thalassemias Hereditary hemolytic diseases in which an imbalance occurs in the synthesis of globin chains The synthesis of either the α- or the β-globin chain is defective Thalassemias are classified into A. β-Thalassemias B. α-Thalassemias

66
β-Thalassemias Synthesis of β-globin chains is decreased or absent (as a result of point mutations) α-Globin chain synthesis is normal. α-Globin tetramers form in RBCs α-Globin tetramers are not stable and precipitate causing the premature death of cells Accumulation of α 2 γ 2 (Hb F) and γ 4 (Hb Bart's) also occurs There are two forms of β-Thalassemia 1. β-Thalassemia major 2. β-Thalassemia minor
 and γ 4 (Hb Bart s) also occurs There are two forms of β-Thalassemia 1. β-Thalassemia major 2. β-Thalassemia minor.")
67
β-Thalassemias β-thalassemia trait (β- thalassemia minor): only one defective β-globin gene β-thalassemia major (Cooley anemia) if both genes are defective Clinical course is different between the two diseases
: only one defective β-globin gene β-thalassemia major (Cooley anemia) if both genes are defective Clinical course is different between the two diseases")
68
β-Thalassemias β-thalassemia minor patients make some β chains; do not require specific treatment β-thalassemia major patients do not develop symptoms until after the first year of life. Why? β-thalassemia major patients require regular transfusions of blood

69
α-Thalassemias Synthesis of α-globin chains is decreased or absent Each individual has four copies of the α-globin gene (two on each chromosome 16) There are several levels of α-globin chain defecencies
 There are several levels of α-globin chain defecencies")
70
One of the four genes is defective, the individual is a silent carrier of α-thalassemia and no physical manifestations of the disease occur If two α-globin genes are defective, the individual has α-thalassemia trait. Why?

71
If three α-globin genes are defective, the individual has hemoglobin H (Hb H) disease, mild to moderate anemia If all four α-globin genes are defective, hydrops fetalis and fetal death result If all four α-globin genes are defective, hydrops fetalis and fetal death result
 disease, mild to moderate anemia If all four α-globin genes are defective, hydrops fetalis and fetal death result If all four α-globin genes are defective, hydrops fetalis and fetal death result")
Similar presentations















 Hb is found in RBCs its main function is to transport O2 to tissues. Structure: 2 parts : heme + globin Globin: four globin chains (2 α.>")

 Chapter 22 Functions of “Blood” Gas Transport Nutrient Transport Excretory Product Transport Cell Signal Transport Hydraulic.>")

 Originally isolated from sperm whales 10X abundance greater in aquatic- than terrestrial-mammals Mb knockout.>")



