Download presentation
Presentation is loading. Please wait.

1
Genetic and developmental disorders II Presented by Ashraf ELMaraghy, MD. Lecturer of Chest Diseases, Ain Shams University

2
Objectives: 1. Developmental causes of upper and lower respiratory tract malformations. 2. Late(adolescent/adult)manifestations of respiratory malformations. 3. Long term sequelae and the residual morbidity of respiratory malformations after management and surgery in infancy and childhood.
manifestations of respiratory malformations. 3. Long term sequelae and the residual morbidity of respiratory malformations after management and surgery in infancy and childhood..")
3
Introduction: There are three ways in which an adult physician may be confronted with this spectrum of disorders. The most frequent type of encounter will be a former pediatric patient, now reaching adulthood, with the history of a surgically treated respiratory malformation; in some of these patients the early loss of lung tissue raises questions of residual damage and compensatory growth. Secondly, there is an increasing number of children in whom pediatric pulmonologists treat respiratory malformations expectantly; these patients eventually become adults with their malformation still in place. Thirdly, there is a small group of patients in whom the malformation goes unrecognized throughout childhood; in these,a late complication or the coincidental discovery of a radiographic anomaly may demand a late diagnosis and management in adulthood.

4
Although congenital lung malformations are rare, they are important disorders because they may lead to considerable morbidity and mortality (e.g., infection, hemorrhage, respiratory failure). Prognosis depends on the size of the lesion and the degree of functional impairment. Small lesions may remain asymptomatic. Failure to recognize a malformation may lead to inappropriate intervention. For example, placement of a chest tube to manage suspected tension pneumothorax in a patient with congenital lobar emphysema may lead to lung contusion and ventilation through the chest tube instead of into the remaining healthy lung.

5
Tracheo-esophageal Fistula (TOF): Pediatric presentation: TOF usually occurs in association with esophageal atresia; thus, the outcome is invariably fatal unless the anomaly is surgically repaired in the first days of life. The rare “H type” TOF without esophageal atresia is an exception to this rule and may persist into later childhood or even adulthood before the diagnosis is established. The malformation is explained by the defective progression of a complex system of folds that separate the foregut into trachea and esophagus. Approximately,50% of infants with TOF have associated anomalies, most often involving the urinary, gastrointestinal and cardiac systems.

7
Long term outcome:Long term outcome: To understand any long term respiratory morbidity after the successful repair of TOF and esophageal atresia, one must realize that the tracheal structure is more or less abnormal in most patients, showing disruption of cartilage rings and a concomitant increase in the membranous portion. This results in tracheomalacia of varying severity which persists far beyond the surgical closure of the TOF and occurs in the majority of patients. In addition, esophageal function tends to remain disturbed after the repair of esophageal atresia ;relevant studies have shown absence of the normal peristaltic wave, atonia and pooling of esophageal contents.

8
The entire spectrum of residual respiratory morbidity after repair of TOF is not sufficiently explained by tracheomalacia alone. In many patients, the remaining abnormalities of esophageal motility cause dysphagia, esophagitis and gastro-esophageal reflux, and these problems tend to persist well into adulthood. There seems to be a correlation between the frequency of bronchitis and pneumonia, on one hand, and the severity of dysphagia on the other. Furthermore, lung function testing in TOF survivors tends to reveal a mildly restrictive respiratory impairment, indicating additional pathology in the lower respiratory tract. All this suggests that some patients tend to aspirate recurrently after the repair of TOF, and this extends the scope of respiratory pathology beyond the malformed trachea.Residual esophageal dysfunction offers itself as the most likely cause for such recurrent aspiration. Scoliosis, which occasionally occurs in patients who have had a TOF repair, is a non-specific long term consequence of thoracotomy.

9
CYSTIC MALFORMATIONS:CYSTIC MALFORMATIONS: Lung cysts: Also termed “Congenital Parenchymal Cysts”. These lesions are a localized malformation of the terminal bronchopulmonary airway. Depending on their origin, their wall may contain bronchial elements such as cartilage, glands and smooth muscle, or they may be of a more bullous alveolar type. Lung cysts may occur as a single or a multicystic lesion, can contain fluid, air, or both, and may or may not communicate with the bronchial tree. The most frequently occurring complication is infection that may enter via rudimentary communications from the airways or via collateral channels, and may lead to protracted pneumonitis and abscess formation.

10
In addition, enlarging cysts may cause compression of the surrounding tissue,resulting in atelectasis and/or recurrent pneumonia. Rupture of a cyst may lead to pneumothorax. Large lesions may cause symptoms postnatally or in infancy but others may remain asymptomatic for years or decades and are only diagnosed by chance or when causing late complications. Diagnostic examination relies on CT scanning; aortography can be used to rule out an aberrant systemic arterial supply to the lesion, thus distinguishing lung cysts from cystic variants of the sequestration spectrum. Surgical resection is usually recommended, especially if the cysts have already caused some complications. Some pediatric pulmonologists will advocate non- surgical management for smaller asymptomatic lesions diagnosed by chance.

13
Cystic adenomatoid malformation (CAM): Cystic adenomatoid malformation is a defect in the development of the terminal bronchioles. A hamartomatous proliferation of cysts occurs and resembles bronchioles (airways without cartilage). Cystic adenomatoid malformation accounts for 25% of all congenital lung malformations. Respiratory distress occurs in the neonatal period, when collateral pores of Kohn ventilate the alveolar tissue present. This process is responsible for the cystic appearance on radiographs. Patients may have mediastinal shift and a pneumothorax. The affected area is dull on percussion and air entry is decreased. The radiographic depiction of a solid or cystic mass on one side of the thorax suggests the diagnosis.
. Cystic adenomatoid malformation accounts for 25% of all congenital lung malformations. Respiratory distress occurs in the neonatal period, when collateral pores of Kohn ventilate the alveolar tissue present. This process is responsible for the cystic appearance on radiographs. Patients may have mediastinal shift and a pneumothorax. The affected area is dull on percussion and air entry is decreased. The radiographic depiction of a solid or cystic mass on one side of the thorax suggests the diagnosis..")
14
Three histological categories of cystic adenomatoid malformation are described: (1) macrocystic (13%), which has the best prognosis and in which one or more large (>5 mm on prenatal ultrasound) cysts are lined with normal pseudostratified ciliated epithelium; (2) microcystic (73%), which has small cysts lined with ciliated columnar or cuboidal epithelium; and (3) solid cystic adenomatoid malformation (13%), which has the worst prognosis and is an airless tissue mass composed of cuboidal epithelium-lined bronchioles. The difference in prognosis may be because the solid and microcystic lesions involve a relatively large amount of lung tissue. Macrocystic lesions are comprised of large, air filled, nonfunctioning spaces involving smaller areas of lungs.

15
Many CAMs present with severe respiratory symptoms at birth; large lesions may cause a mediastinal shift and compression of the opposite lung. Occasionally, it may be difficult to distinguish CAM from congenital diaphragmatic hernia in an infant with severe respiratory distress. However, the diagnosis of some lesions may be delayed until infancy, school age, or even adolescence. These cases present with unresolving pulmonary infiltration, failure to thrive, or pneumothorax. Surgical resection is considered to be the treatment of choice in all cases of CAM. The extent of pulmonary resection will depend on the size of the lesion; some authors advocate surgical techniques that aim to preserve the functional pulmonary tissue that surrounds the malformation.

16
Cystic adenomatoid malformation. Initial radiograph in a patient with congenital cystic adenomatoid malformation on the first day of life with opaque lungs and a suggestion that the right lung is slightly more voluminous than the left lung.

17
Radiograph obtained in the same patient as in the previous image on the second day of life shows that the physiologic fluid is resorbed and replaced with an air-containing cystic area occupying the right upper lung.

18
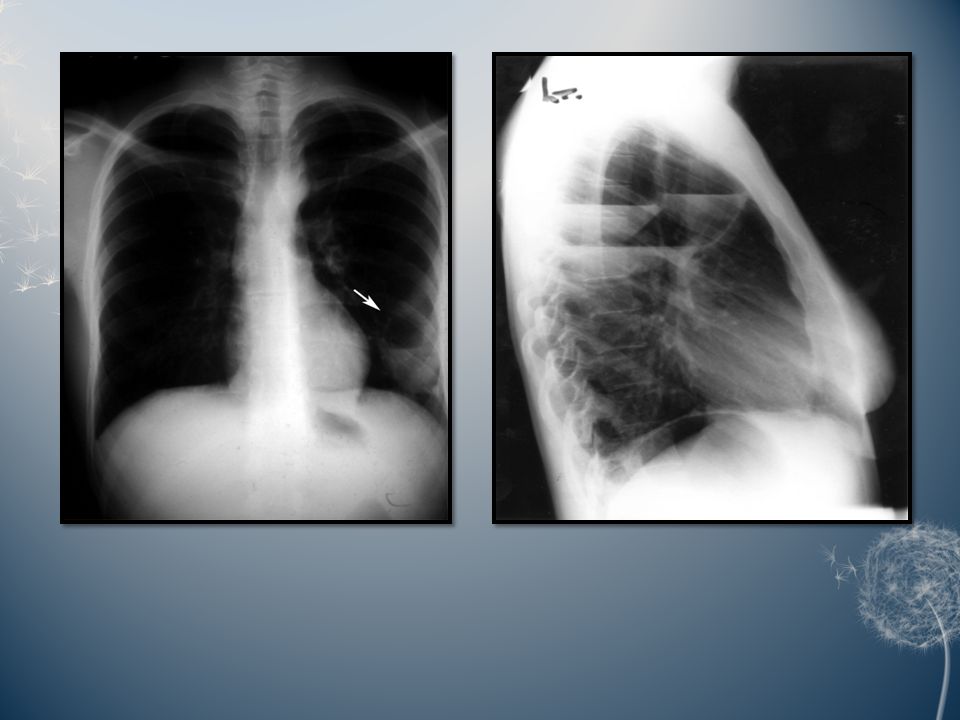
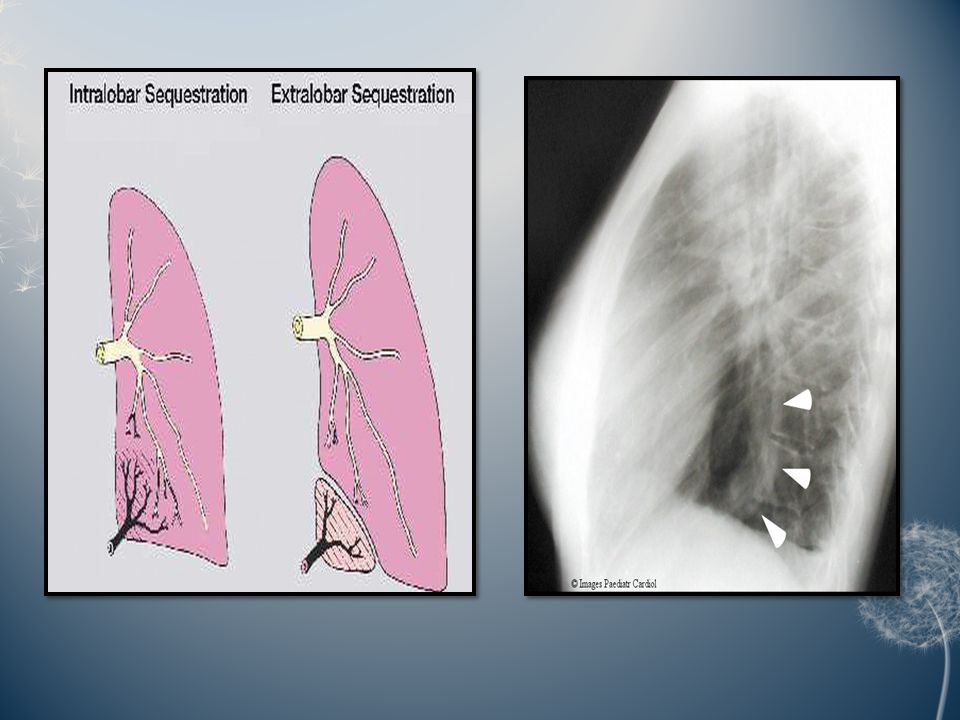
Pulmonary sequestration:Pulmonary sequestration: Pulmonary sequestration is a disconnected or abnormally communicating bronchopulmonary mass or cyst with a normal or anomalous arterial supply and/or venous drainage. It may occur in two forms. In the more frequent intrapulmonary form, the lesion lies within the boundary formed by the pleural layer surrounding the lung, while the rare extra pulmonary form consists of ectopic lung tissue lying outside the boundary formed by the pleural layer that surrounds the rest of the lung. Roughly, two thirds of all pulmonary sequestrations are found in the posterior basal segment of the left lower lobe; the extent of the lesion is usually segmental or less.

19
A bronchial communication is either absent or abnormal, and the lesion may be cystic or not. The aberrant systemic arterial supply arises from the lower thoracic or the upper abdominal aorta or one of its major branches as a single trunk. The venous drainage of a pulmonary sequestration is usually normal to the left atrium, but may also be anomalous to the right atrium, vena cava, or azygos system. Clinically, pulmonary sequestration is latent until infection leads to symptoms. Recurrent pneumonitis of the sequestrated segment, fever, but also purulent sputum and haemoptysis then become the prevailing symptoms. Pulmonary sequestration can present clinically at all ages, but most lesions tend to develop these infective complications at school age and adolescence; however, symptoms may also occur in infants and preschool children, as well as in previously asymptomatic adults.

23
Congenital lobar emphysema (CLE): CLE is a massive postnatal over inflation of one or more lobes of the lung. Approximately, half the cases of CLE involve the left upper lobe and the lesion is less frequently located in the right upper or middle lobe. Congenital heart disease is a common accompaniment of CLE. The development of CLE is still poorly understood. Strictly speaking, the term “emphysema” should be based on clearly defined morphological characteristics that are not present in CLE. This has motivated some authors to suggest the alternative term “congenital lobar over inflation”, but this more accurate nomenclature has never been generally accepted.

24
Some forms of CLE seem to be caused by a bronchial valve mechanism; localized deficiency of bronchial cartilages, bronchial mucosal folds and also extrinsic compression of the bronchus have been described. Another form of CLE might be the result of a true alveolar malformation, as some resected lobes have shown a striking increase in the number of their alveoli. Over distension of the affected lobe may cause compression of the surrounding lung tissue and displacement of the mediastinum. In severe cases, this may manifest as respiratory distress of the newborn. However, later less dramatic manifestations and a coincidental detection of the malformation can occur at any age. In the past, surgical removal of the affected lobe was the standard recommended treatment.

25
However, other reports advocate expectant management and cases of CLE in whom the symptoms gradually resolved have been reported. As a result, most pediatric pulmonologists now try conservative supportive treatment before resorting to surgery. Surgical intervention can thus be reserved for those who do not improve with such a trial and those who present as newborn infants with most severe respiratory embarrassment.

27
Congenital lobar emphysema. Lateral view in the same patient as in the previous image. Same patient as in the previous 2 images. After surgery, the left lung is expanded. A thoracotomy tube is on the right, with a small right-sided pneumothorax.

28
Bronchogenic cysts:Bronchogenic cysts: Bronchogenic cysts are also known as foregut duplication. They arise from an abnormal budding of the ventral foregut. Approximately 85% are mediastinal and 15% are intrapulmonary. The peripheral cysts are multiple and appear late in gestation. They may be filled with air or fluid, or they may have air-fluid levels. The cysts can be central or peripheral. Many are asymptomatic, but incidental findings may be observed on chest radiography. Infection, hemorrhage, and, in rare cases, malignancy can occur. Respiratory distress may result in a stridor or wheeze. Air trapping may lead to emphysema, atelectasis, or both. Dysphagia, chest pain and epigastric discomfort can occur.

29
Bronchogenic cyst. Media file shows a right para tracheal mass. Bronchogenic cyst. Conventional radiographs demonstrate a subcarinal mass.

30
Bronchogenic cyst. CT scan demonstrates a thin-walled cyst in the right upper lobe.

31
Pulmonary agenesis and hypoplasia: Both pulmonary agenesis and hypoplasia may be accompanied by renal anomalies, which are usually apparent soon after birth and associated with respiratory distress. Cardiac defects occur in 50% of patients. Pulmonary agenesis is differentiated from lung aplasia by the absence of the carina in the latter. Lung agenesis is less common than aplasia, about 75% of cases affect the left side, and it is lethal in half of all patients. It may be associated with other manifestations of the syndrome of abnormalities of the vertebrae, anus, cardiovascular tree, trachea, esophagus, renal system and limb buds (VACTERL syndrome). The survival rate is better with left-sided lung agenesis than with right-sided agenesis because the right lung is the larger of the two.
. The survival rate is better with left-sided lung agenesis than with right-sided agenesis because the right lung is the larger of the two..")
32
In pulmonary hypoplasia, development of the distal lung tissue is incomplete. The earlier the delivery of a child, the higher the incidence of lung hypoplasia. In babies delivered before 28 weeks' gestation, the incidence approaches 20%. Pulmonary hypoplasia results from conditions that restrict lung growth, such as oligohydramnios, Potter syndrome (with bilateral renal agenesis or dysplasia), abnormalities of the thoracic cage, Scimitar syndrome (right-sided pulmonary hypoplasia) and diaphragmatic hernia (usually left-sided hypoplasia). More than 50% of patients have associated cardiac, gut, or skeletal malformations. They may have a small thoracic cage, decreased breath sounds on the affected side and a mediastinal shift to the side of the lesion. Therefore, aplasia of the right lung can be confused with dextrocardia. Patients may present with lung infections, dyspnea upon exertion, and/or scoliosis.
, abnormalities of the thoracic cage, Scimitar syndrome (right-sided pulmonary hypoplasia) and diaphragmatic hernia (usually left-sided hypoplasia). More than 50% of patients have associated cardiac, gut, or skeletal malformations. They may have a small thoracic cage, decreased breath sounds on the affected side and a mediastinal shift to the side of the lesion. Therefore, aplasia of the right lung can be confused with dextrocardia. Patients may present with lung infections, dyspnea upon exertion, and/or scoliosis..")
33
Hypovascularity of the entire left lung in a 16-year-old patient with mild exercise intolerance. This patient had hypoplasia of the left lung.

34
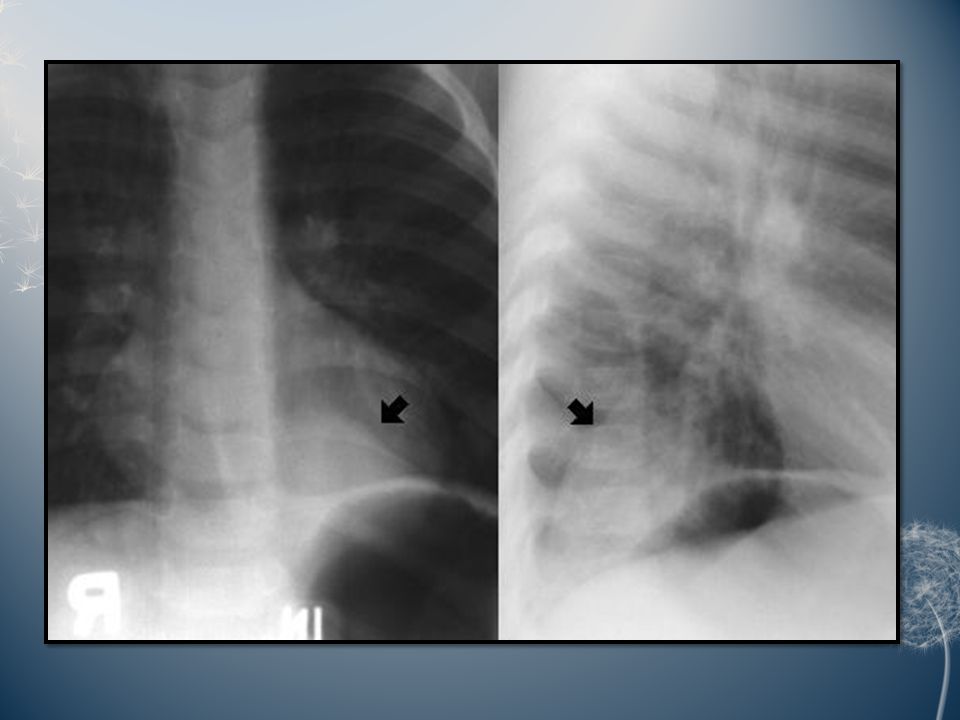
Pulmonary isomerism: Pulmonary isomerism is an anomaly of the number of lung lobes. In the common variety of pulmonary isomerism, the right lung has 2 lobes, whereas the left has 3. This anomaly may be associated with situs inversus, asplenia, polysplenia and/or anomalous pulmonary drainage. Azygous lobe: An azygous lobe is a malformation of the right upper lobe caused by an aberrant azygous vein suspended by a pleural mesentery. An azygous lobe is a radiographic curiosity without clinical significance that occurs in 0.5% of the general population.

37
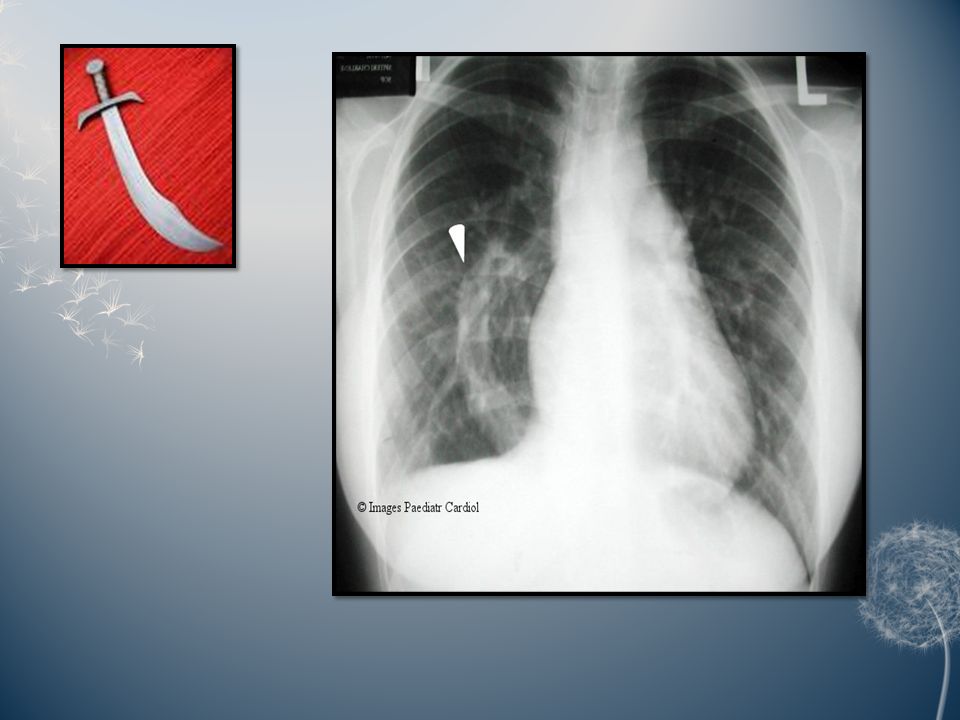
Scimitar syndrome: The constant feature of this syndrome is partial or total anomalous pulmonary venous return to the inferior vena cava. This abnormal vein on the chest radiography creates a gentle curve bulging into the right chest from the mediastinum that some believe resembles the Turkish sword called a scimitar. Other features of the syndrome are variable and may include dextrocardia, hypoplasia of the right lung and/or pulmonary artery, malformation of the bronchi and systemic arterial supply to the right lung. The clinical features vary according to age. Infants almost always present with congestive heart failure and severe pulmonary hypertension. Adults are generally asymptomatic.

40
Hamartoma: Hamartomas are lung nodules contain cartilage, respiratory epithelium and collagen. They may be in the lung tissue or the bronchial lumen. They are presumed to be congenital because they are usually found on chest radiographs in asymptomatic adults. They can cause airway obstruction and are usually excised for diagnosis. Pulmonary lymphangiectasis: Pulmonary lymphangiectasis is a rare disorder in which the normal pulmonary lymphatics are dilated. It may be associated with congenital heart disease in which the pulmonary venous pressure is elevated. Pulmonary lymphangiectasis can also be observed with lymphangiomatosis, in which proliferation of the lymphatic tissue and channels occurs. The disease can also be part of a syndrome of lymphangiomas in many organs; it is sometimes associated with vanishing bones. Pulmonary lymphangiectasis is congenital, but symptoms of respiratory insufficiency usually do not appear until adulthood.

41
Alveolar capillary dysplasia: In alveolar capillary dysplasia, a fatal condition, the distal arteriolar blood supply is reduced, the pulmonary veins are misaligned and the connective tissue between the alveolar epithelium and the capillary endothelium is increased. The alveolar circulation is impaired, and the response to nitric oxide is poor. Affected babies do well with veno-arterial extracorporeal membrane oxygenation (ECMO), but they cannot be weaned from it. The clinical presentation of alveolar capillary dysplasia is that of persistent pulmonary hypertension of the newborn. Hypoxemia leads to arteriolar muscular hypertrophy. Patients may have associated anomalies in the heart or urinary system. Open lung biopsy and cardiac catheterization are suggested as diagnostic tools to look for or exclude pulmonary capillary blush.
, but they cannot be weaned from it. The clinical presentation of alveolar capillary dysplasia is that of persistent pulmonary hypertension of the newborn. Hypoxemia leads to arteriolar muscular hypertrophy. Patients may have associated anomalies in the heart or urinary system. Open lung biopsy and cardiac catheterization are suggested as diagnostic tools to look for or exclude pulmonary capillary blush..")
42
CONGENITAL DIAPHRAGMATIC HERNIA (CDH): Pediatric presentation: CDH is a severe respiratory malformation that occurs with a prevalence of about 0.3 cases per 1000 births. More than 95% of the congenital defects are located in the postero lateral part of the diaphragm; about 85% of these “Bochdalek type” defects are found on the left side. In more than one third of all cases, CDH is associated with other congenital anomalies that involve the cardiovascular, genitourinary, central nervous, or skeletal systems. CDH stems from incomplete development of the septum transversum that normally separates the body cavities between the third and ninth weeks of fetal life. As a result, abdominal viscera are found in the chest and the lungs are hypoplastic.

43
This lung hypoplasia is more severe on the side of the defect; the ipsi lateral lung is small, has reduced size bronchi with less branching, a severely decreased alveolar surface and a pulmonary arterial tree with a reduced number of divisions and an increased thickness of arteriolar walls. The degree of lung hypoplasia is the most critical factor for prognosis, with survival becoming increasingly unlikely in cases where the lung volume deficit approaches 50%. The presence of a near normal lung volume before repair rules out fatal pulmonary hypoplasia and suggests that survival will depend on the successful management of pulmonary hypertension.

45
Long term outcome: Survivors of CDH seem to suffer more severely from viral respiratory infections in infancy and early childhood but, thereafter, most are able to lead a normal life without significant respiratory morbidity and to participate in regular physical activity including sports. Occasionally, however, there is significantly more long term morbidity with failure to thrive and recurrent bronchitis. A few patients only survive with a severe respiratory handicap requiring long term tracheostomy and mechanical ventilation.

46
Chest radiographs after repair of CDH range from normal findings to a radiolucent lung field on the side of the former defect. Pulmonary function tests in CDH survivors tend to show mild to moderate obstructive lung function anomalies. In addition, some patients present with normal pulmonary function tests and some with a restrictive pattern of changes. Many CDH survivors show airway hyper responsiveness when subjected to relevant challenge testing. In contrast to patients with bronchial asthma, however, CDH survivors, while responding positively to pharmacological challenges, do not appear to be hyper responsive to metabisulphite; this suggests that their bronchial hyper responsiveness might stem simply from altered airway geometry. Lung perfusion scans invariably show persisting under perfusion on the side of the former hernia. There is hardly any information on the status of the respiratory pump after neonatal repair of CDH; radiologists have observed an abnormal contour and an impaired motion of the repaired hemidiaphragm.

47
Residual defects in CDH are not restricted to the respiratory system. There is a high prevalence of gastro-esophageal reflux in CDH survivors. Speculatively, this is explained by the frequently occurring congenital absence of the peri- esophageal portion of the diaphragm. Hiatus hernia, susceptibility to intestinal obstruction and recurrence of diaphragmatic hernia, are further components of a long term gastrointestinal morbidity which may be observed after the repair of CDH. The neuro developmental outcome in patients with CDH is comparable to that of other neonates requiring intensive care and extracorporeal membrane oxygenation. Occasionally, there is considerable chronic morbidity from other associated congenital anomalies.

48
Late presentation: Most patients with CDH present within the first 24 hours of life. However, there are a few cases with delayed presentation who develop the first symptoms later in childhood or even as adults. The presenting symptoms are usually gastrointestinal and the ipsi lateral lung is of normal size or only minimally hypoplastic. Late presentation of diaphragmatic hernia is a poorly understood entity; some have challenged the concept of these defects being congenital and have suggested that late presenting diaphragmatic hernias are acquired.

Similar presentations











 of the Lung Dr Bental – NICU – Laniado Hospital בס ד.>")




 Monday, May 04, 2015 Monday, May 04, 2015.>")











 is a common congenital anomaly of the respiratory tract, with an incidence of approximately.>")