Download presentation
Presentation is loading. Please wait.

1
METABOLISM of AMINO ACIDS Dr Shahnaz Khaghani Tehran University of Medical Sciences

2
AMINO ACIDS CATABOLISM pyruvate acetyl-CoA acetoacetate succinyl-CoA, α-KG Anabolic pathways Specialized molecules: creatine, SAM, cysteine, hormones, NO, dTMP ABERRANT METABOLISM PKU, MSUD TRANSAMINATION (Amino nitrogen ) GLUTAMATE UREA
 GLUTAMATE UREA")
3
OUTLINE I.Incorporation of amino acid nitrogen into urea A) Transamination B) Glutamate dehydrogenase generates ammonia C) Urea synthesis requires ammonia, bicarbonate, and ATP 1) hyperammonemia, cause and management II.Catabolism (utilization) of amino acid carbon atoms A) Amino acid carbon atoms pyruvate, thus gluconeogenesis
 Transamination B) Glutamate dehydrogenase generates ammonia C) Urea synthesis requires ammonia, bicarbonate, and ATP 1) hyperammonemia, cause and management II.Catabolism (utilization) of amino acid carbon atoms A) Amino acid carbon atoms pyruvate, thus gluconeogenesis")
4
B) Amino acid carbon atoms acetyl-CoA, thus fatty acids and cholesterol C) Amino acid carbon atoms acetoacetate, thus “ketone bodies” D) Amino acid carbon atoms succinyl-CoA, thus TCA cycle E) Amino acid carbon atoms 2-oxoglutarate (α-KG), thus TCA cycle
 Amino acid carbon atoms acetyl-CoA, thus fatty acids and cholesterol C) Amino acid carbon atoms acetoacetate, thus ketone bodies D) Amino acid carbon atoms succinyl-CoA, thus TCA cycle E) Amino acid carbon atoms 2-oxoglutarate (α-KG), thus TCA cycle")
5
III. Amino acids as substrates in critical anabolic pathways A) Creatine (arginine and glycine) B) Hormones: catecholamines (phenyl- alanine), serotonin (tryptophan), thyroid hormones (phenylalanine) C) “Second messenger,” nitric oxide (NO) (arginine) D) Methyl donor, S-adenosyl-L- methionine or SAM (methionine) E) Cysteine (methionine and serine) F) Methionine resynthesis and DNA synthesis
 Creatine (arginine and glycine) B) Hormones: catecholamines (phenyl- alanine), serotonin (tryptophan), thyroid hormones (phenylalanine) C) Second messenger, nitric oxide (NO) (arginine) D) Methyl donor, S-adenosyl-L- methionine or SAM (methionine) E) Cysteine (methionine and serine) F) Methionine resynthesis and DNA synthesis.")
6
IV. “Inborn errors” of amino acid metabolism A. PKU B. MSUD

7
Amino Acid Breakdown: No storage form of Amino Acids, therefore excess need to be converted to other forms to be used as energy or stored as glycogen/fat. CH2NH2NCH O OH R Disposal of Nitrogen Atom (Urea) Recycling of the Carbon Skeleton
 Recycling of the Carbon Skeleton.")
8
I. Amino Acid Nitrogen Incorporation into Urea A. Transamination Transamination requires: 1) An amino acid and an oxo- (keto-) acid 2) Co-factor pyridoxal phosphate 3) An aminotransferase. NH 2 O O O | | | | | | | R 1 CH– C OH + R 2 C– C– OH O O NH 2 O | | | | | | | R 1 – C – C – O H + R 2 – CH – C – OH
 An amino acid and an oxo- (keto-) acid 2) Co-factor pyridoxal phosphate 3) An aminotransferase. NH 2 O O O | | | | | | | R 1 CH– C OH + R 2 C– C– OH O O NH 2 O | | | | | | | R 1 – C – C – O H + R 2 – CH – C – OH.")
9
-ketoglutarate Aminotransferase B6B6 Flow of Nitrogen: In tissues (e.g Muscle), most amino acids transfer their -amino group to Glutamate
, most amino acids transfer their -amino group to Glutamate")
10
Biosynthesis of Amino Acids: Transaminations 10 Amino Acid 1 + -Keto Acid 2 Amino Acid 2 + -Keto Acid 1 Glutamate + - Ketoglutarate + Pyridoxal phosphate (PLP)- Dependent Aminotransferase
- Dependent Aminotransferase")
11
Transaminations: Role of PLP 11 Tautomerization

12
Aminotransferase The N is then transferred from Glutamate to Pyruvate, producing Alanine. Pyruvate -ketoglutarate

13
Four of the amino acids, Glycine, Lysine, Threonine and Serine are directly deaminated. Serine Dehydratase

14
Glutamine Synthetase [] NH released from Glycine/Lysine/Threonine/Serine is incorporated into Glutamine
![Glutamine Synthetase [] NH released from Glycine/Lysine/Threonine/Serine is incorporated into Glutamine](http://images.slideplayer.com/35/10372798/slides/slide_14.jpg "Glutamine Synthetase [] NH released from Glycine/Lysine/Threonine/Serine is incorporated into Glutamine")
15
In sum: During amino acid breakdown, the -amino Nitrogen gets incorporated as the -amino group in Alanine or the amide group in Glutamine. Alanine and Glutamine are then released to the circulation.

16
Flow of Nitrogen: Alanine and Glutamine released by peripheral tissues are taken up by the Liver. The Nitrogen on Alanine is transferred to -ketoglutarate to produce Glutamate

17
Aminotransferase -ketoglutarate Glutamate

18
Glutamate has two fates important for disposal of waste N. 1)Conversion to -ketoglutarate by Glutamate Dehydrogenase to release NH 3 2) As N donor in the transamination of oxaloacetate to Aspartate
Conversion to -ketoglutarate by Glutamate Dehydrogenase to release NH 3 2) As N donor in the transamination of oxaloacetate to Aspartate.")
19
[ ] Glutamate dehydrogenase 1)Conversion to -ketoglutarate by Glutamate Dehydrogenase to release NH 3
![[ ] Glutamate dehydrogenase 1)Conversion to -ketoglutarate by Glutamate Dehydrogenase to release NH 3](http://images.slideplayer.com/35/10372798/slides/slide_19.jpg "[ ] Glutamate dehydrogenase 1)Conversion to -ketoglutarate by Glutamate Dehydrogenase to release NH 3")
20
2) As N donor in the transamination of oxaloacetate to Aspartate O Glutamate -ketoglutarate NH 2 Oxaloacetate Aspartate Aminotransferase B6B6 OO HO-C-CH 2 -C-C-OH O O HO-C-CH 2 -CH-C-OH
 As N donor in the transamination of oxaloacetate to Aspartate O Glutamate -ketoglutarate NH 2 Oxaloacetate Aspartate Aminotransferase B6B6 OO HO-C-CH 2 -C-C-OH O O HO-C-CH 2 -CH-C-OH")
21
Glutamine is hydrolyzed by Glutaminase to release NH 3 Glutaminase NH

22
Nitrogen flow in Liver: Alanine GlutamateAspartate NH Glutamine

23
The oxoacid can be either pyruvate, which produces alanine, oxaloacetate, which produces aspartate 2-oxoglutarate, which produces glutamate

24
Transamination with 2-oxoglutarate yields glutamate. Glutamate yields ammonia. Ammonia enters the urea cycle. NH 2 O | | | R– CH– C– O H + 2-OG O NH 2 O | | | | | HO– C – CH 2 – CH 2 –CH – C– OH NH 3

25
B. Glutamate dehydrogenase Glutamate to ammonia catalyzed by glutamate dehydrogenase (uses NAD + ) O NH 2 O | | | | | HO–C– CH 2 – CH 2 – CH - C – OH 2-oxoglutarate + NH 3
 O NH 2 O | | | | | HO–C– CH 2 – CH 2 – CH - C – OH 2-oxoglutarate + NH 3.")
27
C. Urea cycle Rate-determining step of the urea cycle requires ammonia, catalyzed by carbamoyl phosphate synthetase. NH 3 + HCO 3 - + 2 ATP CARBAMOYL PHOSPHATE + P i + 2ADP

28
There are two different enzymes with this name. The urea cycle enzyme is carbamoyl phosphate synthetase I or CPS I

29
Carbamoyl phosphate contains nitrogen from ammonia (amino acids), carbon from bicarbonate, and phosphate from ATP. O O │ │ │ │ 2 H N - C - O - P - OH │ OH

30
Next, carbamoyl phosphate reacts with ornithine in a reaction catalyzed by ornithine carbamoyl transferase. Carbamoyl phosphate + ornithine citrulline

31
Net reaction: HCO 3 + NH 3 + 2 ATP (CO 2 + NH 4 ) Carb-Phos + 2 ADP + P i Carbomyl Phosphate Synthetase I
 Carb-Phos + 2 ADP + P i Carbomyl Phosphate Synthetase I")
33
Citrulline reacts with aspartate to yield argininosuccinate. Second nitrogen of urea from aspartate. citrulline + aspartate argininosuccinate synthetase argininosuccinate + AMP + PP i

35
Argininosuccinate is cleaved by arginino- succinate lyase to yield arginine and fumarate. argininosuccinate arginine + fumarate

37
Final reaction, arginine is cleaved by arginase to yield urea and regenerate ornithine (it’s a cycle!) arginine urea + ornithine
 arginine urea + ornithine")
39
CARBAMOYL PHOSPHATE Ornithine Citrulline Argininosuccinate Fumarate Arginine + UREA CYCLE Aspartate +

40
REGULATION OF THE UREA CYCLE Acute: N-acetylglutamate, allosteric effector, up regulates CPS I

41
N-Acetylglutamate is synthesized from glutamate and acetyl-CoA by a mitochondrial NAG synthase.

42
Argininosuccinic acidemia Citrullinuria Arginase Deficiency Hyperammonemia: Type I Type II Metabolic Diseases of the Urea Cycle

43
Disorders present in infants: Symptoms: Lethargy, swelling of the brain leads to mental retardation/brain damage Diagnosis: Low blood urea nitrogen (BUN) levels -high levels of ammonia in the blood elevated circulating glutamine -other metabolites that accumulate depend on the specific enzyme defect Most common form: Hyperammonemia Type II caused by Ornithine Transcarbamylase deficiency Elevated Carb-P levels in this deficiency cause secondary problems in pyrimidine metabolism Metabolic Diseases of the Urea Cycle
 levels -high levels of ammonia in the blood elevated circulating glutamine -other metabolites that accumulate depend on the specific enzyme defect Most common form: Hyperammonemia Type II caused by Ornithine Transcarbamylase deficiency Elevated Carb-P levels in this deficiency cause secondary problems in pyrimidine metabolism Metabolic Diseases of the Urea Cycle")
44
Treatment: Long term, dietary restriction. Low protein diet. Supplemented with Arginine Short term Dialysis Administration of Nitrogen “scavengers” e.g. Phenylacetate

45
Excessive ammonia is toxic to the central nervous system. Alternative pathway therapy: Sodium benzoate to produce hippuric acid Sodium phenylacetate or phenyl- butyrate to produce phenylacetylglutamine

46
NH 3 + CO 2 + 5,10-methylenetetra- hydrofolate NH 2 O | | | Glycine CH 2 – C – OH Sodium Benzoate Bz - N-H O | | | CH 2 – C - OH Benzoyl glycine or hippuric acid excreted

47
NH 3 + glutamate + ATP → glutamine + ADP + P i sodium phenylacetate + CoA → phenylacetyl-CoA Then glutamine + phenylacetyl-CoA → phenylacetylglutamine (excreted)
")
48
sodium phenylbutyrate + CoA → phenylbutyryl-CoA phenylbutyryl-CoA undergoes β-oxidation → phenylacetyl-CoA As above, phenylacetyl-CoA + glutamine → phenylacetylglutamine (excreted)
")
49
II. Catabolic Fates of Amino-acid Carbon Atoms Post-transamination carbon atoms become those of pyruvate acetoacetate acetyl-CoA succinyl-CoA 2-oxoglutarate

50
GLUCOSEPyruvate Oxaloacetate Citrate Fumarate Acetyl CoA Malate -Ketoglutarate Succinyl CoA Succinate Isocitrate Alanine Glutamate Proline Serine Glycine Glutamine Carbon end products from the degradation (catabolism) of amino acids Aspartate Asparagine Amino acids discussed in previous lectures Leucine Tyrosine (50% of carbons) Phenylalanine Cysteine Amino acids to be discussed in this lecture Tyrosine (50% of carbons) Phenylalanine Isoleucine (partial) Valine (partial) Methionine (partial) Methionine (partial)
 of amino acids Aspartate Asparagine Amino acids discussed in previous lectures Leucine Tyrosine (50% of carbons) Phenylalanine Cysteine Amino acids to be discussed in this lecture Tyrosine (50% of carbons) Phenylalanine Isoleucine (partial) Valine (partial) Methionine (partial) Methionine (partial)")
51
Phenylalanine Phenylalanine + O 2 +tetrahydrobiopterin Tyrosine + H 2 O + dihydrobiopterin

52
Critical reactions phenylalanine + O 2 + BH 4 Tyrosine + H 2 O + BH 2

53
Fumarate + Acetyl CoA O2O2 Tyrosine H2OH2O Dihydrobiopterin Phenylalanine hydroxylase Phenylalanine NADP + Dihydropteridine reductase NADPH Tetrahydrobiopterin The phenylalanine hydroxylase reaction and regeneration of its cofactor

54
A. phenylalaninetyrosine Acetoacetate Focus: Degradation of phenylalanine

55
Significance: Acetoacetate is one of the “ketone bodies” and is converted into the other two. Acetoacetate minus CO 2 reduction Acetone 3-Hydroxybutyrate

57
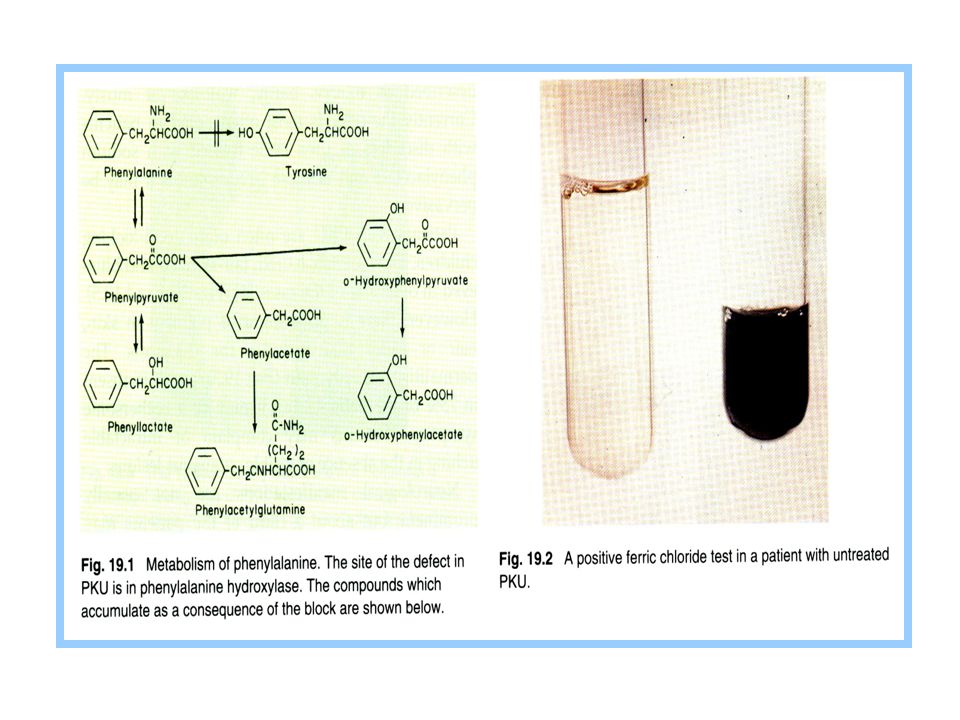
Phenylketonuria (PKU) “Inborn error” in amino acid metabolism is seen when conversion of phenylalanine to tyrosine is blocked. Phenylketones in urine.

58
Phenylketonuria Most common inborn error in amino acid metabolism High phe can cause neurologic damage Unusual compounds: phenylpyruvate; phenyllactate; phenylacetate Brain toxicity: reduced uptake of other aromatic amino acids Tyrosine deficiency may lead to hypopigmentation Cofactor processing can also be defective

59
Phenylketonuria 1) Clinical Manifestations: a) growth failure b) delayed psychomotor development c) seizures d) mental retardation
 Clinical Manifestations: a) growth failure b) delayed psychomotor development c) seizures d) mental retardation")
60
4) Mechanism Hyperphenylalaninemia inhibits amino acid transfer into brain, less dopamine and serotonin 5) Therapies: Low phenylalanine diets
 Mechanism Hyperphenylalaninemia inhibits amino acid transfer into brain, less dopamine and serotonin 5) Therapies: Low phenylalanine diets")
62
Homogentisic Acid Formation Transamination Tyrosine p-Hydroxyphenyl- pyruvate Homogentisate p-Hydroxyphenyl- pyruvate dioxygenase (ascorbate-dep.) O2O2 CO 2 Homogentisate dioxygenase O2O2 Cleavage of aromatic ring Fumarate + acetoacetate Deficient in alkaptonuria
 O2O2 CO 2 Homogentisate dioxygenase O2O2 Cleavage of aromatic ring Fumarate + acetoacetate Deficient in alkaptonuria")
63
Alkaptonuria First defect to which inborn error of metabolism applied – Sir Archibald Garrod in early 1900’s Homogentisate appears in urine Deposited in cartilage and elsewhere polymerization (black) Deficiency of homogentisate dioxygenase Urine turns dark on standing Oxidation of homogentisic acid Asymptomatic in childhood Tendency toward arthritis in adulthood
 Deficiency of homogentisate dioxygenase Urine turns dark on standing Oxidation of homogentisic acid Asymptomatic in childhood Tendency toward arthritis in adulthood")
65
Melanin Formation Highly colored polymeric intermediates Melanin (Black polymer) Tyrosinase DOPA Dopaquinone Tyrosine DOPA OXIDASE Melanin formed in skin (melanocytes), eyes, and hair In skin, protects against sunlight Albinism: genetic deficiency of tyrosinase
 Tyrosinase DOPA Dopaquinone Tyrosine DOPA OXIDASE Melanin formed in skin (melanocytes), eyes, and hair In skin, protects against sunlight Albinism: genetic deficiency of tyrosinase")
66
Albinism Formation of little or no skin pigment Classical defect is tyrosinase (tyrosine hydroxylase)
")
67
Catecholamine Biosynthesis Tyr hydroxylase O2O2 Tyrosine Dihydroxyphenylalanine (DOPA) Dopamine DOPA decarboxylase CO 2 Dopamine hydroxylase Norepinephrine Catechol Epinephrine (Adrenaline) SAM S-Adenosyl- homocysteine Methyl transferase DOPA, dopamine, norepinephrine, and epinephrine are all neurotransmitters
 Dopamine DOPA decarboxylase CO 2 Dopamine hydroxylase Norepinephrine Catechol Epinephrine (Adrenaline) SAM S-Adenosyl- homocysteine Methyl transferase DOPA, dopamine, norepinephrine, and epinephrine are all neurotransmitters")
68
Catechol-O-Methyl Transferase (COMT) COMT Inactive metabolite SAMS-Adenosyl- homocysteine COMT found in cytoplasm Terminates activity of catecholamines Catecholamine excretion products result from combined actions of MAO and COMT Inhibitors of COMT (e.g., tolcapone) useful in Parkinson’s disease Active catecholamine
 COMT Inactive metabolite SAMS-Adenosyl- homocysteine COMT found in cytoplasm Terminates activity of catecholamines Catecholamine excretion products result from combined actions of MAO and COMT Inhibitors of COMT (e.g., tolcapone) useful in Parkinson’s disease Active catecholamine")
69
Tyramine MAO Tyramine found naturally in several types of cheese; also beer and red wine. Tyramine intake can cause hypertensive crisis in persons taking a MAO inhibitor ( norepi release) ( blood pressure)
 ( blood pressure).")
70
Tyrosine BH 4 BH 2 Phenylalanine p-Hydroxyphenylpyruvate HomogentisateMaleylacetoacetate Fumarylacetoacetate L-Dopa Pigmentation Known disorders of phenylalanine and tyrosine metabolism Albinism - tyrosine hydroxylase {tyrosinase} Phenylketonuria – phenylalanine hydroxylase/BH4 synthesis Tyrosinemia type II (Richner-Hanhart) - tyrosine aminotransferase Tyrosinemia type III Tyrosinemia type I Alkaptonuria – homogentisate oxidase
 - tyrosine aminotransferase Tyrosinemia type III Tyrosinemia type I Alkaptonuria – homogentisate oxidase")
71
Tryptophan Metabolism: Biosynthesis of Nicotinic Acid TryptophanNicotinic acid (Niacin) Several steps Nicotinamide adenine dinucleotide (NAD)
 Several steps Nicotinamide adenine dinucleotide (NAD)")
72
Tryptophan Metabolism: Serotonin Formation Tryptophan (Trp) Indole ring Trp hydroxylase O2O2 5-Hydroxy- tryptophan Decarboxylase CO 2 5-Hydroxy- tryptamine (5-HT); Serotonin
 Indole ring Trp hydroxylase O2O2 5-Hydroxy- tryptophan Decarboxylase CO 2 5-Hydroxy- tryptamine (5-HT); Serotonin")
73
Serotonin Metabolism: 5-HIAA Serotonin MAO Dehydrogenase 5-Hydroxyindole acetic acid (5-HIAA) (Urine) Carcinoid tumors: Malignant GI tumor type Excretion of large amounts of 5-HIAA
 (Urine) Carcinoid tumors: Malignant GI tumor type Excretion of large amounts of 5-HIAA")
74
Serotonin Serotonin formed in: Brain (neurotransmitter; regulation of sleep, appetite) Smooth muscle (contraction) Gastrointestinal tract (enterochromaffin cells - major storage site) Drugs affecting serotonin actions used to treat: Depression Serotonin-selective reuptake inhibitors (SSRI) Migraine Schizophrenia
 Smooth muscle (contraction) Gastrointestinal tract (enterochromaffin cells - major storage site) Drugs affecting serotonin actions used to treat: Depression Serotonin-selective reuptake inhibitors (SSRI) Migraine Schizophrenia")
75
Serotonin Metabolism: Melatonin 2 Steps Serotonin Melatonin Melatonin: Formed principally in pineal gland Synthesis controlled by light, among other factors Induces skin lightening Suppresses ovarian function Possible use in sleep disorders

76
Histidine many steps formiminoglutamate tetrahydrofolate 5-formiminotetrahydrofolate ( urine) and glutamate
 and glutamate")
77
Histidine Metabolism: Histamine Formation Histidine Histamine Histidine decarboxylase CO 2 Histamine: Synthesized in and released by mast cells Mediator of allergic response: vasodilation, bronchoconstriction (H 1 receptors) H 1 blockers: Diphenhydramine (Benadryl) Loratidine (Claritin) Stimulates secretion of gastric acid (H 2 receptors) H 2 blockers: Cimetidine (Tagamet); ranitidine (Zantac)
 H 1 blockers: Diphenhydramine (Benadryl) Loratidine (Claritin) Stimulates secretion of gastric acid (H 2 receptors) H 2 blockers: Cimetidine (Tagamet); ranitidine (Zantac)")
78
B. Glycine SerineCysteine AspartateAlanineTryptophan Pyruvate Focus: Catabolism of Glycine

79
NH 2 O | | | CH 2 – C – OH + 5,10-methyleneTHF glycine OH NH 2 O | | | | CH 2 CH – C – OH + THF (non-ligated) serine
 serine")
80
Conversion of Serine to Glycine Folate Tetrahydrofolate (FH 4 ) Dihydrofolate reductase N 5, N 10 -Methylene FH 4 Serine Glycine Serine hydroxymethyl transferase (PLP-dep.) Key intermediate in biosynthesis of purines and formation of thymine Important in biosynthesis of heme, porphyrins, and purines
 Dihydrofolate reductase N 5, N 10 -Methylene FH 4 Serine Glycine Serine hydroxymethyl transferase (PLP-dep.) Key intermediate in biosynthesis of purines and formation of thymine Important in biosynthesis of heme, porphyrins, and purines")
81
OH NH 2 O O O | | | | | | | | CH 2 – CH – C –OH CH 3 – C– C– OH serine pyruvate

82
Tetrahydrofolate is a vehicle for CH 3 - 5-methyl (THF) -CH 2 - 5,10-methylene (THF) -CH= 5,10-methenyl (THF) -C = O 10-formyl (THF) | O - H-C =NH 5-formimino (THF) |
 -CH 2 - 5,10-methylene (THF) -CH= 5,10-methenyl (THF) -C = O 10-formyl (THF) | O - H-C =NH 5-formimino (THF) |")
83
Creatine arginine + glycine guanidino acetic acid S-Adenosyl-L-methionine creatine

84
Methyl donor S-adenosyl-L-methionine (SAM) from methionine methionine + ATP S-adenosyl-L-methionine + PP i + P i
 from methionine methionine + ATP S-adenosyl-L-methionine + PP i + P i")
85
SAM: required methyl donor for creatine, epinephrine, phosphatidylcholine, and for DNA methylation.

86
Creatine and Creatinine Arginine Glycine Ornithine Arginine-glycine transamidinase (Kidney) Guanidoacetate Methyltransferase (Liver) SAM + ATP S-Adenosyl- homocysteine + ADP Creatinine (Urine) Non-enzymatic (Muscle) Creatine kinase (Muscle) ATP Creatine Phosphocreatine ADP + Pi
 Guanidoacetate Methyltransferase (Liver) SAM + ATP S-Adenosyl- homocysteine + ADP Creatinine (Urine) Non-enzymatic (Muscle) Creatine kinase (Muscle) ATP Creatine Phosphocreatine ADP + Pi")
87
Creatine and Creatinine Creatine: Dietary supplement Used to improve athletic performance Creatinine: Urinary excretion generally constant; proportional to muscle mass Creatinine Clearance Test: Compares the level of creatinine in urine (24 hrs.) with the creatinine level in the blood Used to assess kidney function Important determinant in dosing of several drugs in patients with impaired renal function
 with the creatinine level in the blood Used to assess kidney function Important determinant in dosing of several drugs in patients with impaired renal function")
88
c. LeucineIsoleucine Lysine Acetyl-CoA Focus: Catabolism of leucine and isoleucine

89
D. IsoleucineValine Methionine Threonine Succinyl- CoA Focus: Propionyl-CoA Succinyl-CoA

90
Propionyl-CoA converted into succinyl-CoA via a vitamin B 12 - dependent rearrangement. propionyl-CoA methylmalonyl-CoA methylmalonyl-CoA mutase vitamin B 12 succinyl-CoA

91
Significance: Carbon atoms of succinyl-CoA TCA cycle

92
Leucine Isoleucine Valine Branched-chain -ketoacid CO 2 + NADH Glutamate -Ketoglutarate Transaminase CoA + NAD + Branched-chain ketoacid dehydrogenase CoA derivatives of each branched-chain ketoacid 3 Acetyl CoA from leucine biotin Methylmalonyl CoA Propionyl CoA biotin Succinyl CoA adenosyl B 12 multiple enzyme reactions Pathways for the catabolism of leucine, isoleucine and valine

93
Rate-determining step (RDS) catalyzed by Branched-chain oxoacid dehydrogenase complex (BCOD complex).
 catalyzed by Branched-chain oxoacid dehydrogenase complex (BCOD complex).")
94
MAPLE SYRUP URINE DISEASE Most common BCAA disorder (1/185,000) Defective branched-chain ketoacid dehydrogenase Similar to PDH with 3 enzyme activities Thiamine deficiency can produce same result Keto acids that accumulate smell like burn maple syrup BCAA also accumulate Mental retardation Untreated leads to death
 Defective branched-chain ketoacid dehydrogenase Similar to PDH with 3 enzyme activities Thiamine deficiency can produce same result Keto acids that accumulate smell like burn maple syrup BCAA also accumulate Mental retardation Untreated leads to death")
95
B. Maple syrup urine disease (MSUD) 1) Clinical observations (About 80%) neonatal onset with severe encephalopathy and coma; (about 20%) milder disease
 1) Clinical observations (About 80%) neonatal onset with severe encephalopathy and coma; (about 20%) milder disease.")
96
2) Critical reaction BCOD complex oxidation of transamination product 3) Critical enzyme BCOD complex 4) Mechanism of disease Acute encephalopathy, coma due to “keto-leucine”
 Critical reaction BCOD complex oxidation of transamination product 3) Critical enzyme BCOD complex 4) Mechanism of disease Acute encephalopathy, coma due to keto-leucine")
97
In adolescents and young adults, dysmyelination due to chronically increased leucine 5) Therapy Drugs (e.g., clofibrate) that decrease activity of BCOD kinase (low activity of BCOD complex as effective as possible)
 Therapy Drugs (e.g., clofibrate) that decrease activity of BCOD kinase (low activity of BCOD complex as effective as possible)")
Similar presentations
















 Urea cycle and reactions that feed amino groups into the cycle. The enzymes catalyzing these reactions (named in the text) are distributed.>")









>")