Download presentation
Presentation is loading. Please wait.

1
Hepatobiliary Disorders
Dr. Mohamed Hesham Sayed Professor of Pediatrics Rabigh Faculty of Medicine, KAU

2
Objectives Define and classify causes of cholestasis.
Describe the consequence of cholestasis Correlate the abnormalities in LFT with cholestasis. Recognize feature of biliary atresia. Discuss viral hepatitis Recognize feature and causes of chronic liver disease. Describe end stage of liver disease

8
Clinical presentations of liver diseases
1) Hepatomegaly Infections: viral hepatitis, abscess, Bilharziasis Congestive: CHF, veno-occlusive disease Metabolic : Glycogen storage, Wilson disease Malignant : liver tumors, neuroblastoma 2) Jaundice Hemolytic anemias Acute and chronic liver diseases 3) Portal hypertension Collateral circulation(abd veins &varices) Splenomegaly Ascites
 Hepatomegaly. Infections: viral hepatitis, abscess, Bilharziasis. Congestive: CHF, veno-occlusive disease. Metabolic : Glycogen storage, Wilson disease. Malignant : liver tumors, neuroblastoma. 2) Jaundice. Hemolytic anemias. Acute and chronic liver diseases. 3) Portal hypertension. Collateral circulation(abd veins &varices) Splenomegaly. Ascites.")
9
4) Acute liver cell failure
Progressive jaundice Bleeding Rapidly developing coma 5) Chronic liver cell failure General: anorexia, wt loss Jaundice Ascites Bleeding Skin : palmar erythema & spider nevi Encephalopathy
 Chronic liver cell failure. General: anorexia, wt loss. Jaundice. Ascites. Bleeding. Skin : palmar erythema & spider nevi. Encephalopathy.")
11
Reasons for a Delay in Referral of Infants Who Have Liver Disease
Lack of follow-up of neonatal jaundice (including failure to fractionate serum bilirubin) Inadequate investigation of hemorrhagic disease/ coagulopathy Misdiagnosis of cholestasis (conjugated hyperbilirubinemia) as human milk jaundice (unconjugated hyperbilirubinemia) False security due to a fall in serum bilirubin concentrations or presence of pigmented stools
 Inadequate investigation of hemorrhagic disease/ coagulopathy. Misdiagnosis of cholestasis (conjugated hyperbilirubinemia) as human milk jaundice (unconjugated hyperbilirubinemia) False security due to a fall in serum bilirubin concentrations or presence of pigmented stools.")
12
Causes of Liver Diseases
The causes of liver disease in pediatric patients vary with age: Neonates and infants Biliary atresia and idiopathic neonatal hepatitis older children/adolescents Acetaminophen intoxication and Wilson disease

13
Causes of Liver Disease in Pediatric Patients According to Age Neonates and Infants
Cholestatic disorders —Biliary atresia —Choledochal cyst —Paucity of intrahepatic bile ducts (eg, Alagille syndrome) —Progressive familial intrahepatic cholestasis syndromes (Byler disease and syndrome) Idiopathic neonatal hepatitis and mimickers —Cystic fibrosis —Alpha 1-antitrypsin deficiency
 —Progressive familial intrahepatic cholestasis. syndromes (Byler disease and syndrome) Idiopathic neonatal hepatitis and mimickers. —Cystic fibrosis. —Alpha 1-antitrypsin deficiency.")
14
Viral hepatitis or other infectious diseases
—Cytomegalovirus —Herpes simplex virus/herpes zoster virus/human herpesvirus 6 Metabolic disease Tyrosinemia, galactosemia, fructosemia Others Total parenteral nutrition

15
Causes of Liver Disease in Pediatric Patients According to Age Older Children and Adolescents
Hepatitis —Viral hepatitis (hepatitis B virus, hepatitis C virus) —Autoimmune hepatitis Wilson disease Liver disease associated with inflammatory bowel disease Parasitic infections Toxins and pharmacologic remedies Malignancies Portal hypertension Fatty liver of obesity (nonalcoholic steatohepatitis) Hypotension/ischemia/cardiac failure
 —Autoimmune hepatitis. Wilson disease. Liver disease associated with inflammatory bowel disease. Parasitic infections. Toxins and pharmacologic remedies. Malignancies. Portal hypertension. Fatty liver of obesity (nonalcoholic steatohepatitis) Hypotension/ischemia/cardiac failure.")
16
Neonatal Cholestasis

17
beyond the first 14 days of life
Definition Failure of bile to reach duodenum due to Liver or biliary disorder Prolonged elevation of serum conjugated bilirubin beyond the first 14 days of life

19
Causes of cholestasis 1) Idiopathic neonatal hepatitis
The commonest cause due to unknown etiology 2) Infections : Bacterial, viral or protozoal 3) Metabolic - Carbohydrate (galactosemia), - Amino acids (tyrosinemia), - Others (alpha-1-antitrypsin deficiency) 4) Bile duct obstruction: Extrahepatic biliary atresia (EHBA), Choledochal cyst 5) Familial syndromes as paucity of intrahepatic bile ducts (Allagile syndrome)
 Infections : Bacterial, viral or protozoal. 3) Metabolic. - Carbohydrate (galactosemia), - Amino acids (tyrosinemia), - Others (alpha-1-antitrypsin deficiency) 4) Bile duct obstruction: Extrahepatic biliary atresia (EHBA), Choledochal cyst. 5) Familial syndromes as paucity of intrahepatic bile ducts (Allagile syndrome)")
20
Allagile syndrome

21
Consequences of cholestasis
1) Decreased bile excretion - Fat malabsorption - Pale or clay colored stool 2) Retention of bile - Jaundice - Pruritis - Progressive liver damage
 Decreased bile excretion. - Fat malabsorption. - Pale or clay colored stool. 2) Retention of bile. - Jaundice. - Pruritis. - Progressive liver damage.")
22
Clinical features * The diagnosis is considered in case of
- Persistent neonatal jaundice > 2 wks - Hepatomegaly, - Pale stools, and - Dark urine * Signs of the cause may include - Low birth weight (TORCH), - Cataract (galactosemia or rubella), or - Abnormal facies in Allagile syndrome
, - Cataract (galactosemia or rubella), or. - Abnormal facies in Allagile syndrome.")
23
1) α1-antitrypsin deficiency
 α1-antitrypsin deficiency")
24
Neonatal Cholestasis - History
Jaundice in any infant after 2 weeks of age should raise the suspicion of liver disease and prompt appropriate evaluation. Onset related to diet….inborn error/metabolism Family history…. Alagille syndrome, IEM Male/LBW….neonatal hepatitis Female……EHBA Early onset/acholic stool….EHBA

25
Goals of timely evaluation
Diagnose and treat known medical and/or life-threatening conditions. Identify disorders amenable to surgical therapy within an appropriate time-frame. Avoid surgical intervention in intrahepatic diseases.

26
Rx- Kasai portoenterostomy
Surgical correction of biliary atresia ideally should occur within the first 2 months of life. Rx- Kasai portoenterostomy Age < 8wks 90% clear jaundice Age 8-12 wks 45% Age>12wks 22%

27
Rapid diagnosis of biliary atresia

28
Extrahepatic biliary atresia
Generally acholic stools with onset at about 2 weeks-old Average birth weight Hepatomegaly with firm to hard consistency Female predominance No well-documented familial cases

29
Extrahepatic biliary atresia
Increased incidence of polysplenia syndrome and intra-abdominal vascular anomalies Normal uptake on radionucleotide scan with absent excretion Biopsy shows bile duct proliferation, bile plugs, portal or perilobular fibrosis and edema, and intact lobular structure

30
Idiopathic neonatal hepatitis
Generally normal stools or acholic stools with onset at one month-old Low birth weight Normal liver on exam or hepatomegaly with normal to firm consistency Male predominance Familial cases (15-20%)
")
31
Idiopathic neonatal hepatitis
Impaired uptake on radionucleotide scan with normal excretion Biopsy shows intralobular inflammation with focal hepatocellular necrosis and disruption of the hepatic architecture. No alteration of the bile ducts. Giant cell transformation occurs but is non-specific.

32
Infantile Cholestasis - Objectives
Aim 1: Identify a treatable cause Aim 2: Recognise complications Aim 3: Early referral to specialist unit when necessary

33
Etiologies Basic distinction is between: Extrahepatic etiologies
Intrahepatic etiologies

34
Extrahepatic etiologies
Extrahepatic biliary atresia Choledochal cyst Bile duct stenosis Spontaneous perforation of the bile duct Cholelithiasis Inspissated bile/mucus plug Extrinsic compression of the bile duct

35
Intrahepatic etiologies
Idiopathic Neonatal Hepatitis Toxic TPN-associated cholestasis Drug-induced cholestasis Genetic/Chromosomal Infectious Metabolic Miscellaneous

37
Presentation Jaundice- conjugated bilirubin Pale stools Dark urine
Abnormal LFTs - high or low GGT Bleeding Hepatosplenomegaly Abdominal mass Failure to thrive

38
Investigations of cholestasis
A) To document diagnosis of cholestasis - Total and direct serum bilirubin - Liver enzymes: ALT (more liver specific), AST, alkaline phosphatase and gamma glutamyl transpeptidase - Serum proteins (total and albumin) - Prothrombin time and concentration
 To document diagnosis of cholestasis - Total and direct serum bilirubin - Liver enzymes: ALT (more liver specific), AST, alkaline phosphatase and gamma glutamyl transpeptidase - Serum proteins (total and albumin) - Prothrombin time and concentration")
39
B) To define the cause of cholestasis
1) Look for treatable conditions - Galactosemia (reducing substance in urine); - Septicemia (sepsis screen) 2) Look for congenital infections - TORCH screenig 3) Look for metabolic conditions - Tyrosinemia and alpha one antitrypsin deficiency 4) Look for choledochal cyst : by abdominal US
 Look for treatable conditions. - Galactosemia (reducing substance in urine); - Septicemia (sepsis screen) 2) Look for congenital infections. - TORCH screenig. 3) Look for metabolic conditions. - Tyrosinemia and alpha one antitrypsin deficiency. 4) Look for choledochal cyst : by abdominal US.")
43
5) Differentiate between idiopathic hepatitis and extrahepatic atresia: A) Radionuclide scanning (HIDA scan) - If dye in intestine hepatitis - If no dye in intestine EHBA B) Liver biopsy Most helpful but not 100% conclusive - Giant cell transformation hepatitis - Fibrosis of portal tracts/bile duct proliferation EHBA C) Operative cholangiogram - To demonstrate patency or obstruction of bile ducts
 Differentiate between idiopathic hepatitis and extrahepatic atresia: A) Radionuclide scanning (HIDA scan) - If dye in intestine hepatitis - If no dye in intestine EHBA B) Liver biopsy Most helpful but not 100% conclusive - Giant cell transformation hepatitis - Fibrosis of portal tracts/bile duct proliferation EHBA C) Operative cholangiogram - To demonstrate patency or obstruction of bile ducts")
44
Biliary Atresia Radioisotope scan (TBIDA) of liver showing good hepatic uptake of isotope and no excretion into bowel.
 of liver showing good hepatic uptake of isotope and no excretion into bowel.")
45
Biliary Atresia Liver biopsy of biliary atresia showing bands of fibrous tissue with bile duct proliferation.

46
Neonatal Hepatitis Liver biopsy in neonatal hepatitis showing inflammatory infiltrate throughout the liver, and giant cell and rosette formation of liver cells.

47
Biliary cirrhosis

48
Biliary cirrhosis

49
Kasai Portoenterostomy
Formation of Roux-en-Y to re establish bile flow by anastomising bowel to ductal remnant

50
Management of cholestasis
1) Specific treatment - Sepsis antibiotics - Galactosemia Lactose free milk - Choledochal cyst surgical correction - EHBA Kasai operation (hepatic portoenterostomy) Should be done before 60 days to obtain good results as the obliterative process is progressive Delay in operation reduces the chances to find any patent biliary ductules. Procedure: a loop of jejunum is anastomosed to the transected porta hepatis.
 Specific treatment - Sepsis antibiotics - Galactosemia Lactose free milk - Choledochal cyst surgical correction - EHBA Kasai operation (hepatic portoenterostomy) Should be done before 60 days to obtain good results as the obliterative process is progressive Delay in operation reduces the chances to find any patent biliary ductules. Procedure: a loop of jejunum is anastomosed to the transected porta hepatis.")
52
2) Nutritional support Medium chain triglycerides and fat soluble vitamins 3) Treatment of complications - Pruritis: ursodeoxycholic acid or bile acid binders as cholestyramine - Varices: sclerotherapy - Hepatic encephalopathy: 10% glucose infusion, bowel wash by enema, oral neomycin, lactulose,…. 4) Liver transplantation - For EHBA who have failed Kasai.
 Liver transplantation. - For EHBA who have failed Kasai.")
53
Cholestasis in Older Children
HAV infection (anorexia, fever, vomiting, abdominal pain, darkening of the urine with elevated aminotransferase values in the absence of other known hepatotoxic exposures). HCV infection (increased exposure to IV blood products (hemodialysis, hemophilia, surgery). Hepatotoxic medications(isoniazid, nitrofurantoin, sulfonamides, and NSAID , such as acetaminophen and ibuprofen.
. HCV infection (increased exposure to IV blood products (hemodialysis, hemophilia, surgery). Hepatotoxic medications(isoniazid, nitrofurantoin, sulfonamides, and NSAID , such as acetaminophen and ibuprofen.")
54
Cholestasis in Older children
A history of sore throat in an individual who also has jaundice, splenomegaly, and lymphadenopathy suggests Epstein-Barr virus. Gall bladder disease, right upper quadrant colicky pain and nausea (following ingestion of fatty foods). Cholestasis may lead to complaints of pruritus and a particularly dark and foamy urine. The color is due to choluria (bile pigments in the urine); the foaminess suggests the presence of choleuria (bile salts in the urine).
. Cholestasis may lead to complaints of pruritus and a particularly dark and foamy urine. The color is due to choluria (bile pigments in the urine); the foaminess suggests the presence of choleuria (bile salts in the urine).")
55
Viral Hepatitis HAV HBV HCV HDV HEV Type RNA DNA RNA, incomplete
Transmission Oral Parenteral Incubation, day 15-40 50-150 30-150 20- 90 15-60 Carrier state No yes Diagnosis Anti-HAV IgM HBsAg, Anti-HBcIgM Anti-HCV, HCV-RNA Anti-HDV Anti-HEV Vaccine Yes Treatment Not needed Interferon-alpha/ Lamivudine Ribavirin Interferon-alpha

56
Diagnosis of viral hepatitis

57
1) Clinical forms of acute hepatitis (4 forms)
a) Icteric hepatitis: the most common Pre - icteric phase : - Mild fever , anorexia , vomiting and abdominal pain - Urine is dark ( bilirubinuria ) Icteric phase : - Jaundice appears - Liver is tender and enlarged - Anorexia is common Convalescent phase : - Gradual decline of symptoms and signs. - This is the rule in hepatitis A.
 Icteric hepatitis: the most common Pre - icteric phase : - Mild fever , anorexia , vomiting and abdominal pain - Urine is dark ( bilirubinuria ) Icteric phase : - Jaundice appears - Liver is tender and enlarged - Anorexia is common Convalescent phase : - Gradual decline of symptoms and signs. - This is the rule in hepatitis A.")
58
b) Cholestatic hepatitis obstruction to bile flow, jaundice, and pruritis and pale stool are common
c) Anicteric hepatitis - Common in infants featured by GIT manifestations like gastroenteritis d) Fulminant hepatitis - Not common and very serious - Causing acute liver failure (progressive jaundice, bleeding and rapidly developing coma)
 Anicteric hepatitis. - Common in infants featured by GIT manifestations like gastroenteritis. d) Fulminant hepatitis. - Not common and very serious. - Causing acute liver failure (progressive jaundice, bleeding and rapidly developing coma)")
59
2) Investigation to confirm acute liver injury
- Marked increase of serum bilirubin (direct or mixed) - Marked elevations of transaminases (ALT and AST) to very high levels. - Bilirubin in urine - Evidence of acute hepatic failure Rising serum bilirubin Low serum albumin Prolonged prothrombin time High blood ammonia, and electrolyte imbalance
 - Marked elevations of transaminases (ALT and AST) to very high levels. - Bilirubin in urine. - Evidence of acute hepatic failure. Rising serum bilirubin. Low serum albumin. Prolonged prothrombin time. High blood ammonia, and electrolyte imbalance.")
60
3) Identification of the virus
HAV HBV HCV HDV HEV Onset Acute Insidious More insidious With HBV As HAV Course Short prolonged More prolonged Severe Chronicity No possible Very possible Fulminant failure Rare May occur rare In pregnant females Laboratory diagnosis Anti-HAV IgM (acute), Anti-HAVIgG (recovery) HBsAg/ antiHBc IgM (acute), Anti-HBs (recovery), antiHBc IgG (chronicity) Anti-HCV (exposure), Anti-HCV/ HCVRNA (chronic) Anti-HCV and –ve HCVRNA
, Anti-HAVIgG. (recovery) HBsAg/ antiHBc IgM (acute), Anti-HBs (recovery), antiHBc IgG. (chronicity) Anti-HCV (exposure), Anti-HCV/ HCVRNA. (chronic) Anti-HCV and –ve HCVRNA.")
61
Prevention Hepatitis A - Fecal - oral hygiene - Hepatitis A vaccine Hepatitis B - Measures for blood products (use disposable syringes and needles), screening of pregnant mothers, immunization - Passive : gammaglobulin - Active : hepatitis B vaccine.
, screening of pregnant mothers, immunization. - Passive : gammaglobulin. - Active : hepatitis B vaccine.")
62
Complications : in types B,C,D:
(1) Fulminant hepatitis & hepatic failure (2) Chronic hepatitis ( persistent - active ) (3) Cirrhosis (4) Hepatocellular carcinoma (5) Aplastic anemia
 Fulminant hepatitis & hepatic failure. (2) Chronic hepatitis ( persistent - active ) (3) Cirrhosis. (4) Hepatocellular carcinoma. (5) Aplastic anemia.")
63
Acute liver failure (fulminant hepatitis)
Massive hepatic necrosis with subsequent loss of liver function, Uncommon, but has a high mortality. The child may present within hours or weeks with jaundice, encephalopathy, coagulopathy, hypoglycaemia and electrolyte disturbance. Early signs of encephalopathy include alternate periods of irritability and confusion with drowsiness. Older children may be aggressive and unusually difficult. Complications include cerebral oedema, haemorrhage from gastritis or coagulopathy, sepsis and pancreatitis.

64
Acute liver failure (fulminant hepatitis)
Causes Infection…HAV, HBV, HCV, non-A to G Drugs/Poisons…Paracetamol, isoniazide,… Metabolic…Wilsons disease, tyrosinemia… Autoimmune hepatitis Reye’s syndrome

65
Acute liver failure (fulminant hepatitis)
Management Maintaining the blood glucose (>4mmol/L) by IV dextrose. preventing sepsis with broad-spectrum antibiotics preventing haemorrhage IV vitamin K, FFP and H2-blockers treating cerebral oedema by fluid restriction and mannitol diuresis.
 by IV dextrose. preventing sepsis with broad-spectrum antibiotics. preventing haemorrhage IV vitamin K, FFP and H2-blockers. treating cerebral oedema by fluid restriction and mannitol diuresis.")
66
Acute liver failure (fulminant hepatitis)
Poor prognosis is likely when the liver begins to shrink in size, if there is a rising bilirubin with falling transaminases, an increasing coagulopathy or progression to coma. Without liver transplantation, 70% of children who progress to coma will die.

67
Reye's syndrome and Reye-like syndrome
Acute non-inflammatory encephalopathy with microvesicular fatty infiltration of the liver. Aetiology is unknown, there is a close association with aspirin therapy. Stop give aspirin to children as an antipyretic to avoid it Reye-like syndrome Beta oxidation defect, medium chain acyl-CoA dehydrogenase deficiency (MCAD). Many of these patients would have presented with acute liver failure and a Reye-like syndrome later in life.
. Many of these patients would have presented with acute liver failure and a Reye-like syndrome later in life.")
68
Chronic hepatitis

69
Causes 1) Chronic infection: HBV, HDV, and HCV (not with HAV or HEV) 2) Autoimmune hepatitis 3) Drug-induced: Rifampicin, isoniazid, nitrofurantoin 4) Inborn errors of metabolism As Wilson disease and alpha-one anti-trypsin deficiency
 Chronic infection: HBV, HDV, and HCV (not with HAV or HEV) 2) Autoimmune hepatitis 3) Drug-induced: Rifampicin, isoniazid, nitrofurantoin 4) Inborn errors of metabolism As Wilson disease and alpha-one anti-trypsin deficiency")
71
Autoimmune Hepatitis The mean age is 7-10 years. More common in girls.
May present as an acute hepatitis, as fulminant hepatic failure or chronic liver disease with autoimmune features such as skin rash, lupus erythematosus, arthritis, haemolytic anaemia or nephritis. Diagnosis is based on hypergammaglobulinaemia (IgG >20g/L); positive autoantibodies, e.g. smooth muscle antibodies (SMAs), antinuclear antibodies (ANAs) or liver/kidney microsomal antibodies. a low serum complement (C4); and typical histology. Most children respond to prednisolone and azathioprine.
; positive autoantibodies, e.g. smooth muscle antibodies (SMAs), antinuclear antibodies (ANAs) or liver/kidney microsomal antibodies. a low serum complement (C4); and typical histology. Most children respond to prednisolone and azathioprine.")
72
Wilson’s Disease Kayser-Fleischer rings from copper in the cornea in a child with Wilson's disease.

73
Clinical presentations
1) Unresolved hepatitis Acute hepatitis unresolving in 6 months 2) Slowly progressive liver disease Manifestations of chronic liver cell failure
 Unresolved hepatitis. Acute hepatitis unresolving in 6 months. 2) Slowly progressive liver disease. Manifestations of chronic liver cell failure.")
74
Investigations 1. Liver functions tests: usually abnormal 2. Abdominal ultrasound: to define size of liver and spleen and check for ascites 3. Liver biopsy: is very helpful for diagnosis, it shows chronic inflammatory cell infiltrate 4. Investigations for the cause such as serological markers for hepatitis B and C; autoantibodies for autoimmune hepatitis and low seruloplasmin and high copper in Wilson disease.

75
Complications Liver cirrhosis and hepatic carcinoma
Portal hypertension causing varices and ascites Chronic liver cell failure

76
Treatment 1) Treatment of cause if possible
Interferon for chronic HBV and HCV Prednisolone and azathioprine for autoimmune hepatitis D- penicillamine for Wilson disease 2) Liver transplantation Good option for end stage liver disease with good results
 Liver transplantation. Good option for end stage liver disease with good results.")
77
Liver Cirrhosis Definition : Irreversible condition with widespread liver fibrosis & nodule formation. There is disturbed vascular arrangement leading to portal hypertension Causes : 1 - Post-hepatitis cirrhosis 2 - Biliary cirrhosis eg. biliary atresia 3 - Metabolic e.g. Wilson's disease . 4 - Cardiovascular: Constrictive pericarditis- Budd chiari synd. Diffuse fibrosis occur in : - Schistosomiasis Cong. hepatic fibrosis

78
Complications (1) Parenchymal Failure (liver call failure) * Failure of synthetic function of liver - Decrease S. albumin, prothrombin, coagulation factors * Failure of detoxication A - encephalopathy B - hormonal changes C - infection (2) Vascular failure i.e. Portal hypertension Splenomegaly, Ascites and Portosystemic anastomosis (oesophageal varices)
 Parenchymal Failure (liver call failure) * Failure of synthetic function of liver - Decrease S. albumin, prothrombin, coagulation factors * Failure of detoxication A - encephalopathy B - hormonal changes C - infection (2) Vascular failure i.e. Portal hypertension Splenomegaly, Ascites and Portosystemic anastomosis (oesophageal varices)")
79
Portal hypertension Definition: When the portal venous pressure exceeds 12 mmHg (normal variation 5 – 10 mmHg) Causes of portal hypertension: 1) Pre hepatic = portal vein obstruction/thrombosis due to - umbilical sepsis or - umbilical vein catheterization * Clinically: 1 - Portal hypertension 2 - Marked splenic enlargement with NORMAL liver
 Pre hepatic = portal vein obstruction/thrombosis due to. - umbilical sepsis or - umbilical vein catheterization. * Clinically: 1 - Portal hypertension. 2 - Marked splenic enlargement with NORMAL liver.")
80
2) Intra hepatic portal hypertension : Hepatic - Chronic hepatitis - Cirrhosis - Bilharzial fibrosis - Wilson disease - Congenital hepatic fibrosis - Veno-occlusive disease Biliary - Extrahepatic biliary atresia - Sclerosing cholangitis * Clinically : - History of hepatic/biliary disease - Portal hypertension - Liver size is variable, usually shrunken with sharp edge.
 Intra hepatic portal hypertension : Hepatic - Chronic hepatitis - Cirrhosis - Bilharzial fibrosis - Wilson disease - Congenital hepatic fibrosis - Veno-occlusive disease Biliary - Extrahepatic biliary atresia - Sclerosing cholangitis * Clinically : - History of hepatic/biliary disease - Portal hypertension - Liver size is variable, usually shrunken with sharp edge.")
81
3) Post hepatic portal hypertension : (postsinusoidal) - Budd-Chiari ( hepatic vein obstruction) - Constrictive pericarditis . * Clinically : - Portal hypertension - Marked liver enlargement - Spleen may be normal or enlarged in late cases

82
Clinical features 1) Esophageal varices: due to opening of collateral circulation. Also, dilated abdominal veins (caput medusa). Varices lead to hematemesis which may be mild or massive. 2) Splenomegaly: More evident with prehepatic and hepatic causes 3) Ascites: Due to - Hypoproteinemia, - Na retention, - Renal impairment and fluid redistribution . It can be complicated by bacterial peritonitis.
 Esophageal varices: due to opening of collateral circulation. Also, dilated abdominal veins (caput medusa). Varices lead to hematemesis which may be mild or massive. 2) Splenomegaly: More evident with prehepatic and hepatic causes 3) Ascites: Due to - Hypoproteinemia, - Na retention, - Renal impairment and fluid redistribution . It can be complicated by bacterial peritonitis.")
83
4) Hepatic encephalopathy: induced by GIT hemorrhage, sepsis, electrolyte imbalance and high protein intake. The mechanisms of CNS manifestations include hyperammonemia, false neurotransmitters, high aromatic amino acid levels and GABA accumulation 5) Renal failure: hepatorenal syndrome.
 Renal failure: hepatorenal syndrome.")
84
Cirrhosis and Portal Hypertension
(i) malnutrition with loss of fat and muscle bulk (ii) distended abdomen from ascites and hepatosplenomegaly (iii) scrotal swelling from ascites (iv) no jaundice despite advanced liver disease.
 malnutrition with loss of fat and muscle bulk. (ii) distended abdomen from ascites and hepatosplenomegaly. (iii) scrotal swelling from ascites. (iv) no jaundice despite advanced liver disease.")
85
Ascites This infant has A grossly distended abdomen from ascites
Dilated abdominal veins secondary to portal hypertension and An umbilical hernia from increased abdominal pressure A surgical scar.

86
Cirrhosis and Portal Hypertension

87
Cirrhosis and Portal Hypertension

88
Portal Hypertension, Varices

89
Portal Hypertension, Varices

90
Portal Hypertension, Varices

91
Investigations 1) Upper endoscopy: to detect varices
2) Abdominal ultrasound: may show shrunken liver and splenomegaly 3) Splenoportography to locate site of obstruction 4) Liver function tests 5) Screening for causes of chronic liver disorders
 Abdominal ultrasound: may show shrunken liver and splenomegaly. 3) Splenoportography to locate site of obstruction. 4) Liver function tests. 5) Screening for causes of chronic liver disorders.")
92
Management Aim: to prevent or treat bleeding varices
A) Emergency treatment of bleeding varices 1. Hospitalization 2. IV fluid resuscitation and blood transfusion 3. H2 blockers as ranitidine 4. If bleeding persists: vasopressin infusion
 Emergency treatment of bleeding varices. 1. Hospitalization. 2. IV fluid resuscitation and blood transfusion. 3. H2 blockers as ranitidine. 4. If bleeding persists: vasopressin infusion.")
93
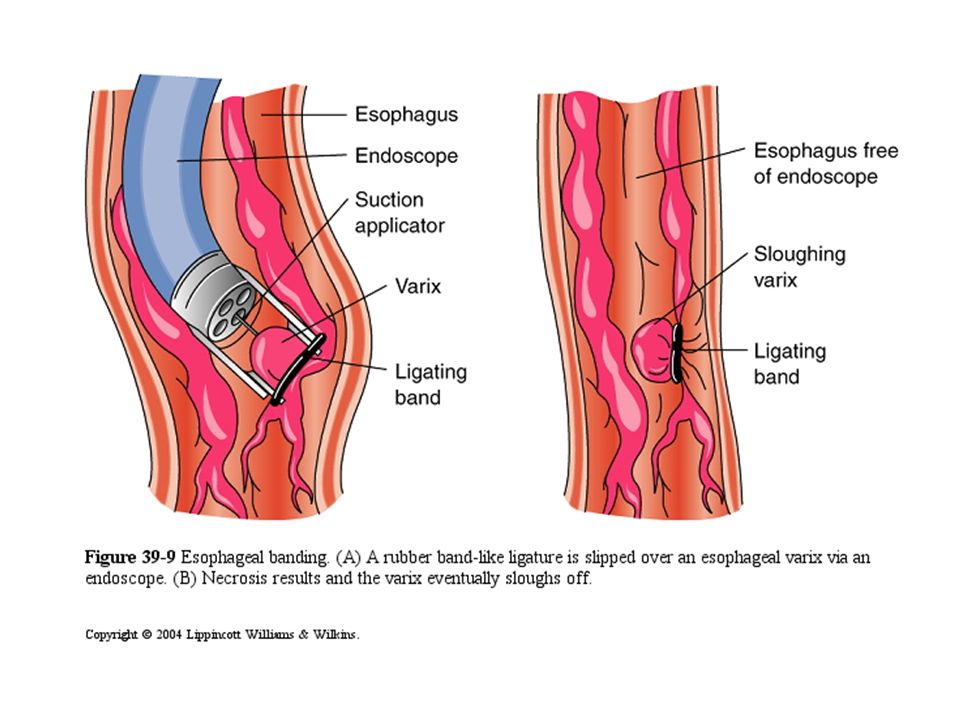
B) Endoscopic management by - Sclerotherapy or - Band ligation of the varices C) Surgical treatment by portosystemic shunt (bypass) - Transjugular intrahepatic porto-systemic shunt (TIPSS) - Surgical portosystemic shunts as porticaval shunts
 Endoscopic management by - Sclerotherapy or - Band ligation of the varices C) Surgical treatment by portosystemic shunt (bypass) - Transjugular intrahepatic porto-systemic shunt (TIPSS) - Surgical portosystemic shunts as porticaval shunts")
97
D) Prevention of bleeding
- Prevention of first bleeding attack: Avoid aspirin Beta blockers as propranolol(reduces portal venous pressure) prophylactic sclerotherapy or band ligation - Prevention of rebleeding: beta blockers, sclerotherapy, band ligation, surgical portosystemic shunts and liver transplantation
 prophylactic sclerotherapy or band ligation. - Prevention of rebleeding: beta blockers, sclerotherapy, band ligation, surgical portosystemic shunts and liver transplantation.")
98
Liver transplantation
Indications include acute or chronic end stage liver failure. Contraindications include sepsis, untreatable cardiopulmonary disease or cerebrovascular disease. Most children receive part of an adult’s liver of a living person.

99
Complications after transplantation are not uncommon such as hepatic artery thrombosis, biliary leaks, rejection, VOD and sepsis Outcome: Children who survive the initial postoperative period usually do well. Short and long term results are much promising for this kind of therapy for previously hopeless cases.

100
Clinical and laboratory evaluation of liver disease

101
Physical Examination The liver span is the distance between the liver edge and the upper margin of dullness obtained by percussion at the right midclavicular line. The mean span changes from 4.5 to 5 cm at 1 week of age to 6 to 7 cm in early adolescence.

102
If the spleen is enlarged: Tender hepatomegaly: Massive hepatomegaly
Portal hypertension or storage disease Tender hepatomegaly: Viral hepatitis or heart failure Massive hepatomegaly Congenital hepatic fibrosis Ascites: Portal hypertension or liver failure Massive hepatosplenomegaly: Storage disorder or malignancy

103
Intense pruritus in obstructive liver disease, that primarily is manifested by irritability.
Alagille syndrome Characteristic facies (beaked nose, high forehead), butterfly vertebrae, a murmur on auscultation due to peripheral pulmonic stenosis, and a posterior embryotoxon on ophthalmologic examination.
, butterfly vertebrae, a murmur on auscultation due to peripheral pulmonic stenosis, and a posterior embryotoxon on ophthalmologic examination.")
104
Laboratory Evaluation
Cholestatic or obstructive Liver cell injury Elevation of mainly alkaline phosphatase (AP), gamma glutamyl transpeptidase (GGT), and conjugated bilirubin. Elevation of mainly aminotransferases (ALT and AST)
, gamma glutamyl transpeptidase (GGT), and conjugated bilirubin. Elevation of mainly aminotransferases (ALT and AST)")
105
Liver function tests The term is NOT accurate… Why??
Only two of the parameters commonly obtained are true measures of hepatic function the prothrombin time (PT) and serum albumin level, both assess synthetic ability. All of the other parameters are essentially indirect measures of liver function, and some of these values are altered in settings other than liver disease
 and. serum albumin level, both assess synthetic ability. All of the other parameters are essentially indirect measures of liver function, and some of these values are altered in settings other than liver disease.")
106
Liver function tests Cholestasis is defined by conjugated hyperbilirubinemia more than 20 % of total serum bilirubin. Dark urine due to bilirubinuria The levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) are the most sensitive tests of hepatocyte necrosis. ALT is more specific of liver disease because it is found only in low concentrations in other tissues .
 and aspartate aminotransferase (AST) are the most sensitive tests of hepatocyte necrosis. ALT is more specific of liver disease because it is found only in low concentrations in other tissues .")
107
Liver function tests An elevated serum AP level usually indicates obstructive liver disease especially if the rise in AP is associated with a rise in GGT… Why?? A low serum albumin concentration is a late finding in liver disease. When it is present, it suggests chronic disease. Hyperammonemia and encephalopathy are classic findings of liver failure, and there is a labile correlation between the degree of encephalopathy and serum ammonia levels.

108
Liver function tests The PT (prothrombin time) which measures the time required for prothrombin (factor II) to be converted into thrombin, will be elevated in the presence of biliary obstruction. This is the basis for parenteral administration (not oral) of vitamin K to patients who have elevated PT values.
 which measures the time required for prothrombin (factor II) to be converted into thrombin, will be elevated in the presence of biliary obstruction. This is the basis for parenteral administration (not oral) of vitamin K to patients who have elevated PT values.")
109
Liver function tests No matter what the cause, an elevated PT value in patients who have liver disease is serious. One of the first steps when evaluating a newborn who has cholestasis is to measure PT/PTT and administer vitamin K. Untreated hypoprothrombinemia may lead to spontaneous bleeding and intracranial hemorrhage.

110
Imaging and Histopathology of the Liver and Biliary Tract
Abdominal ultrasonography Liver size and texture Choledochal cysts and stones (95% accuracy) Space occupying lesions & dilated bile ducts. Absence of gallbladder in case of biliary atresia. Radionuclide imaging (technetium-99) Detects liver uptake and excretory abilities. The appearance of the tracer within the intestinal region by 24 hours virtually excludes biliary atresia, but the converse is not true.
 Space occupying lesions & dilated bile ducts. Absence of gallbladder in case of biliary atresia. Radionuclide imaging (technetium-99) Detects liver uptake and excretory abilities. The appearance of the tracer within the intestinal region by 24 hours virtually excludes biliary atresia, but the converse is not true.")
111
Percutaneous liver biopsy
The cardinal method by which to arrive quickly at a diagnosis of underlying liver disease. Shows the degree of fibrosis/cirrhosis and permits the diagnosis of biliary atresia, neonatal hepatitis, congenital hepatic fibrosis, and alpha 1-antitrypsin deficiency. In neonatal hepatitis there is giant cell transformation with inflammatory infiltrates of the portal zones and an absence of bile ductule proliferation.

112
Percutaneous liver biopsy
In EHBA there is proliferation of the interlobular bile ducts, periportal fibrosis, and bile plugs in canaliculi and ductules.

113
Older children Wilson disease needs to be included in the differential diagnosis of any child who presents with liver disease, neurologic abnormalities, behavioral changes, or Kayser- Fleischer rings.

114
Management

115
Liver transplantation
Survival rates approach 80% at 1 year and 70% at 5 years. Biliary atresia is the most common indication for transplant and may be the initial treatment when detected late or may be used as a salvage procedure for a failed Kasai. Used early in cases of tyrosinemia

116
Medical management of liver diseases
Nutritional support Treatment of pruritus, choleretics and bile acid-binders Management of portal hypertension and its consequences

117
Treatment Nutritional support Adequate calories and protein
Supplement calories with medium chain triglycerides Treatment and/or prophylaxis for fat-soluble vitamin deficiencies (vitamins A, D, E, and K)
")
118
Treatment Nutritional support (cont.)
Supplemental calcium and phosphate when bone disease is present Prophylaxis for zinc deficiency Low-copper diet as poorly excreted Sodium restriction when ascites present

119
Treatment Treatment of pruritus Bile acid-binders: cholestyramine
Ursodeoxycholic acid Phenobarbital as a choleretic

120
Treatment Management of portal hypertension and its consequences
Variceal bleeding Fluid resuscitation Blood products Sclerotherapy Balloon tamponade Portovenous shunting Propanolol

121
Treatment Management of portal hypertension and its consequences (cont.) Ascites Sodium restriction Diuretics: spironolactone, furosemide Albumin Paracentesis

122
Conclusion Identifying the presence of serious liver disease in a pediatric patient at the initial presentation is of cardinal importance. Early recognition of babies who have biliary atresia is critical for optimal medical or surgical intervention.

123
Case Studies

124
A 15-year-old girl who is being evaluated for poor school performance and acting-out behaviors is noted to have a large, firm liver. Kayser- Fleischer rings are apparent on ophthalmologic examination. Determination of which of the following is most likely to confirm the diagnosis in this patient? . A. Hepatic copper concentration. B. Hepatic iron concentration. C. Hepatitis B antibody titers. D. Serum alpha 1-antitrypsin level. E. Serum chloride level

125
A 3-year-old girl who has cholestasis and is malnourished is suspected of having a nutrient deficiency. Which one of the following clinical findings is most likely to be present in this child? . A. Megaloblastic anemia. B. Neuritis. C. Pellagra. D. Photophobia. E. Rachitic rosary

126
Thank you

Similar presentations



















>")

>")

 LIVER FUNCTION AND THE BILIARY TRACT LECTURE FOUR Dr. Essam H. Aljiffri.>")
 that progresses to cirrhosis Replacement of liver tissue.>")
 LIVER FUNCTION AND THE BILIARY TRACT LECTURE FIVE Dr. Essam H. Aljiffri.>")








